CDK4 INHIBITOR
Neuroblastoma is a malignant pediatric tumor with a wide range of stages, requiring a wide range of therapeutic options. However, successful therapeutic options remain limited. Therefore, it is urgently necessary to identify additional chemotherapeutic agents to target this disease. It has been reported that artemisinin has multiple anti-proliferative activity, including cell growth suppression (26,27), apoptosis induction (28), angiogenesis inhibition, cell migration disruption (29–31), and modulation of nuclear receptor responsiveness (32,33). Yet, the effect of artemisinin on neuroblastoma remains unclear. The results presented in the present study demonstrated that artemisinin led to significantly decreased cell growth and cell proliferation, and increased apoptosis in neuroblastoma cells. We first demonstrated that artemisinin treatment suppressed the ability of colony formation in vitro and tumorigenicity of neuroblastoma cells in vivo.
Artemisinin has been suggested to promote cytostasis by G0/G1-phase arrest and to decrease the expression level of cyclinB1, CDK2 and CDC25A in colon cancer (34), and to inhibit the promoter activity of CDK4 in prostate cancer (16). Artemisinin also induced G2/M phase arrest in osteosarcoma cells (35). Our data indicated that artemisinin induced cell cycle arrest at the G1 phase, together with a decrease in the expression levels of cyclinD1, CDK4 and cyclinE2 in all four neuroblastoma cell lines. cyclinB1 was downregulated only in the BE(2)-C and SHEP1 cells, but no significant difference was noted in the SK-N-AS and SK-N-DZ cells.
Previous studies have shown that artemisinin induced apoptosis in pancreatic tumor cells (36), lung adenocarcinoma cells (37), liver cancer cells (38), and non-small cell lung cancer cells (39). In the present study, we first demonstrated that artemisinin induced the cell death and apoptosis of neuroblastoma cells at a lower dose than that for clinical usage. Next, we investigated the mechanism and the expression of apoptotic relevant proteins.
Collectively, our results revealed that artemisinin inhibited cell proliferation and tumor growth, with cell cycle arrest and apoptosis induction in neuroblastoma cells. Since artemisinin has been used for the treatment of malaria for an extensive period of time, a large body of data regarding clinical tests and adverse drug reactions in patients are available. Therefore, artemisinin may serve as a potential new therapeutic agent for the treatment of neuroblastoma.
Artocarpin
NOXA ACTIVATOR
In conclusion, our results demonstrated that whereas artocarpin induces cytotoxic effects in human NSCLC cells, it may exert protective effects in normal HPAEpiCs. In addition, artocarpin may serve as a pro-oxidant only in human NSCLC cells, but not in normal HPAEpiCs. Previously, various flavonoids (including flavone acetic acid, quercetin, and flavopiridol) have entered human clinical trials, and have shown promising anticancer effects clinically [54]. We propose that cell apoptosis caused by artocarpin-induced oxidative stress and ROS generation can be an important mechanism for cancer prevention and therapy. Additionally, this study is the first to demonstrate that artocarpin-induced apoptosis is mediated through activation of the Nox2/p47phox pathway leading to enhanced ROS production, which then induces the activation of two distinct signaling cascades, including ERK MAPK/ p38 MAPK/p53-dependent activation of PUMA/Cytochrome C/ Apaf-1/ caspase 3 pathway in A549 cells and PI3K/ Akts473/ p53-independent activation of NF-kB/ c-Myc/Noxa pathway in both A549 and H1299 cells
P53 Activator
The transcription factor NF-kB is a major PI3K/Akt downstream effector, and plays a dual role as an attenuator or promoter of apoptosis. It regulates the transcription of DNA, and mediates apoptosis in response to oxidative stress [38]. Recent studies have demonstrated that ROS-dependent NF-kB activation induced the protein expression of c-Myc and Noxa in p53-independent human NSCLC cell death [50]. On the contrary, NF-kB activation was involved in resistance to oxidative stress and p53-mediated programmed cell death [51–53]. Therefore, we investigated whether this transcription factor may play a role in artocarpin-induced apoptosis. In the current study, we observed that artocarpin induced the activation of NF-kB via a Nox2/ROS/PI3K/Akt dependent signaling pathway. Correspondingly, amelioration of Nox2/ROS/PI3K/Akt pathway significantly attenuated artocarpin-induced translocation ofNF-kB and up-regulation of c-Myc and Noxa proteins. These results demonstrated that activation of the p53-independent ROS/NF-kB/c-Myc/Noxa signaling pathway by artocarpin plays a critical role in inducing apoptosis in A549 and H1299 cells.
Aspalathin
NRF2 Activator
Aspalathin (ASP) can protect H9c2 cardiomyocytes against high glucose (HG)-induced shifts in myocardial substrate preference, oxidative stress, and apoptosis. The protective mechanism of ASP remains unknown. However, as one of possible, it is well known that phytochemical flavonoids reduce oxidative stress via nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activation resulting in up-regulation of antioxidant genes and enzymes. Therefore, we hypothesized that ASP protects the myocardium against HG- and hyperglycemia-induced oxidative damage by up-regulating Nrf2expression in H9c2 cardiomyocytes and diabetic (db/db) mice, respectively. Using an oxidative stress RT2 Profiler PCR array, ASP at a dose of 1 μM was demonstrated to protect H9c2 cardiomyocytes against HG-induced oxidative stress, but silencing of Nrf2 abolished this protective response of ASP and exacerbated cardiomyocyte apoptosis. Db/db mice and their non-diabetic (db/+) littermate controls were subsequently treated daily for six weeks with either a low (13 mg/kg) or high (130 mg/kg) ASP dose. Compared to nondiabetic mice the db/db mice presented increased cardiac remodeling and enlarged left ventricular wall that occurred concomitant to enhanced oxidative stress. Daily treatment of mice with ASP at a dose of 130 mg/kg for six weeks was more effective at reversing complications than both a low dose ASP or metformin, eliciting enhanced expression of Nrf2 and its downstream antioxidant genes. These results indicate that ASP maintains cellular homeostasis and protects the myocardium against hyperglycemia-induced oxidative stress through activation of Nrf2and its downstream target genes.

Astragalus complanatus seed extract
Bax, P21, P27 levels increased / cyclinD1, CDK1, and CDK4 levels decreased.
Aim of the study: Flavonoids extracted from the seeds of Astragalus complanatus R.Br. reduce the proliferation of many cancer cells. The present study was carried out to evaluate the effects of these flavonoids fromAstragalus complanatus (FAC) on human hepatocarcinoma cell viability and apoptosis and to investigate its mechanisms of action in SMMC-7721 cells.
Materials and methods: Cell viability was measured using the MTT assay. To detect apoptotic cells, SMMC- 7721 cells treated with FAC were stained with Hoechst 33258 and subjected to agarose gel electrophoresis. Quantitative detection of apoptotic cells was performed by flow cytometry. The effects of FAC on apoptosis and cell cycle regulatory genes and proteins in SMMC-7721 cells were examined using an S series apoptosis and cell cycle gene array and Western blot analysis.
Results: The growth of SMMC-7721 and HepG2 cells was inhibited by treatment with FAC. Cell death induced by FAC was characterized by nuclear condensation and DNA fragmentation. Moreover, the cell cycle was arrested in the G0/G1 and S phases in FAC-treated SMMC-7721 cells. A sub-G1 peak with reduced DNA content was also formed. The activity of caspase-3 was significantly increased following FAC treatment. Microarray data indicated that the expression levels of 76 genes were changed in SMMC-7721 cells treated with FAC: 35 genes were up-regulated and 41 were down-regulated. Western blot analysis showed that caspase-3, caspase-8, Bax, P21, and P27 protein levels in SMMC-7721 cells were increased after 48 h of FAC treatment, while cyclinB1, cyclinD1, CDK1, and CDK4 protein levels were decreased.Conclusions: These results suggest that FAC may play an important role in tumor growth suppression by inducing apoptosis in human hepatocarcinoma cells via mitochondria-dependent and death receptor- dependent apoptotic pathways.
Avicin D
StAT3, BCL-2, VEGF Inhibitor
Avicins, a class of electrophilic triterpenoids with pro-apoptotic, anti-inflammatory and antioxidant properties, have been shown to induce redox-dependant post-translational modification of cysteine residues to regulate protein function. Based on (a) the cross-talk that occurs between redox and phosphorylation processes, and (b) the role of Stat3 in the process of apoptosis and carcinogenesis, we chose to study the effects of avicins on the processes of phosphorylation/dephosphorylation in Stat3. Avicins dephosphorylate Stat3 in a variety of human tumor cell lines, leading to a decrease in the transcriptional activity of Stat3. The expression of Stat3-regulated proteins such as c-myc, cyclin D1, Bcl2, survivin and VEGF were reduced in response to avicin treatment. Underlying avicin-induced dephosphorylation of Stat3 was dephosphorylation of JAKs, as well as activation of protein phosphatase-1. Downregulation of both Stat3 activity and expression of Stat 3-controlled pro-survival proteins, contributes to the induction of apoptosis in avicin treated tumor cells. Based on the role of Stat3 in inflammation and wounding, and the in vivo inhibition of VEGF by avicins in a mouse skin carcinogenesis model, it is likely that avicin-induced inhibition of Stat3 activity results in the suppression of the pro-inflammatory and pro-oxidant stromal environment of tumors. Activation of PP-1, which also acts as a cellular economizer, combined with the redox regulation by avicins, can aid in redirecting metabolism from growth promoting anabolic to energy sparing pathways.
Increases caspase-3 and -8; inhibits PI3K/Akt, NFƙB, iNOS, and MMP-2 and -9; attenuates Bcl-2; induces Bax
The inhibitory effect of baicalein, which is present in Indian trumpet flower and Chinese skullcap, or Scutellaria baicalensis, on human colon cancer was studied in vitro and in vivo 85. Research indicated that baicalein had a significant inhibitory effect on HCT-116 cells. The mechanisms of effect of baicalein occur through three pathways: i) the extrinsic pathway of apoptosis, ii) by decreasing the incidence of inflammation, and iii) by impairment of tumor formation through inactivation of the PI3K/Akt pathway. Baicalein increased the expression of caspase-3 and -8, which are involved in apoptosis. The expression of NF-ƙB was inhibited, resulting in inhibition of iNOS, MMP-9, and MMP-2 genes, all of which are involved in inflammation 86, 87. The effect of baicalein on HT-29 cells was also investigated. The data indicated that baicalein had the ability to increase cell arrest in the G1 phase. Baicalein attenuated the expression of Bcl-2, whereas the expression of Bax was augmented. Moreover, induction of apoptosis was achieved by inactivation of the PI3K/Akt pathway 88. Further studies on cancerous Institute for Cancer Research (ICR) mice induced by AOM supported the preventive effect of baicalein 86.
► Scutellaria baicalensis (SB) and SB-derived polyphenols possess anti-proliferative activities in pancreatic cancer. ► Baicalein decreased mRNA and protein expression of the anti-apoptotic Bcl-2 family protein Mcl-1 and induced apoptosis. ► Mcl-1 knock-down induced apoptosis through caspase cascade, but Bcl-2 or Bcl-xL knock-down had no or only a slight effect. ► The effect of baicalein on apoptosis was significantly attenuated by Mcl-1 over-expression.
BAVACHIN
( from Psoralea corylifolia)
Bavachin induces the apoptosis of multiple myeloma cell lines by inhibiting the activation of nuclear factor kappa B and signal transducer and activator of transcription 3
Bavachin is a phytoestrogen purified from natural herbal plants such as Psoralea corylifolia. In this study, we examined the effect of bavachin in multiple myeloma(MM) cell lines. We found that bavachin decreased the viability of MM cell lines, but was not cytotoxic towards normal cells. It inhibited the activation of nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3). Furthermore, bavachin increased the expression of p53 and NOXA, and decreased the expression of X-linked inhibitor of apoptosis protein (XIAP), survivin, B cell lymphoma-extra large (Bcl-xL), and Bcl-2.Additionally, bavachin induced apoptosis by the activation of caspase-3 and caspase-9, implicating the involvement of the mitochondrial pathway. Our results suggest that bavachin induces apoptosis through the inhibition of NF-κB and STAT3 activation in MM cell lines. Most importantly, few NF-κB and STAT3 inhibitors with high efficiency, specificity, and safety are currently available for clinical cancer therapy. Hence, bavachin, which targets NF-κB and STAT3, is a potential anticancer agent for the treatment of MM.
Berberine
(from Berberis Aristata aka “Indian Barberry”, “Chutro” and “Tree Turmeric”)
Key Functions:
Inhibits HIF-1a
These data indicated that HIF-1α repression is a critical step in the inhibitory effect of berberine on tumor-induced angiogenesis. Northern blot analyses plus pulse-chase assays revealed that berberine did not down-regulate HIF-1α mRNA but destabilized HIF-1α protein. We found that berberine-induced HIF-1α degradation was blocked by a 26S proteasome inhibitor. Moreover, immunoprecipitation and Western blot analyses showed that berberine increased the lysine-acetylated HIF-1α in hypoxic SC-M1 cultures. These data indicated that a proteasomal proteolytic pathway and lysine acetylation were involved in berberine-triggered HIF-1α degradation. In conclusion, our data provided molecular evidence to support berberine as a potent antiangiogenic agent in cancer therapy
Attenuation of premature cellular senescence
Aging is the greatest risk factor for human diseases, as it results in cellular growth arrest, impaired tissue function and metabolism, ultimately impacting life span. Two different mechanisms are thought to be primary causes of aging. One is cumulative DNA damage induced by a perpetuating cycle of oxidative stress; the other is nutrient-sensing adenosine monophosphate-activated protein kinase (AMPK) and rapamycin (mTOR)/ ribosomal protein S6 (rpS6) pathways. As the main bioactive component of natural Chinese medicine rhizoma coptidis (RC), berberine has recently been reported to expand life span in Drosophila melanogaster, and attenuate premature cellular senescence. Most components of RCincluding berberine, coptisine, palmatine, and jatrorrhizine have been found to have beneficial effects on hyperlipidemia, hyperglycemia and hypertension aging-related diseases. The mechanism of these effects involves multiple cellular kinase and signaling pathways, including anti-oxidation, activation of AMPK signaling and its downstream targets, including mTOR/rpS6, Sirtuin1/ forkhead box transcription factor O3 (FOXO3), nuclear factor erythroid-2 related factor-2 (Nrf2), nicotinamide adenine dinucleotide (NAD+) and nuclear factor-κB (NF-κB) pathways. Most of these mechanisms converge on AMPK regulation on mitochondrial oxidative stress. Therefore, such evidence supports the possibility that rhizoma coptidis, in particular berberine, is a promising anti-aging natural product, and has pharmaceutical potential in combating aging-related diseases via anti-oxidation and AMPK cellular kinase activation.
Suppresses Activation of pi3k/AKT Signaling
Berberine (BBR), an isoquinoline alkaloid originally isolated from the Chinese herb Coptis chinensis (Huanglian), exhibits anti‑inflammatory and immunosuppressive properties. Since myocardial ischemia/reperfusion (I/R) injury is associated with an excessive immune response, the current study was conducted to investigate the impact of BBR on myocardial I/R injury, a common disorder in clinical settings. Preconditioning of Sprague‑Dawley rats with BBR (100 mg/kg/day, by gavage) for 14 days prior to the induction of I/R significantly attenuated myocardial I/R injury as manifested by a reduction in the incidence of ventricular arrhythmia and the amelioration of myocardial histological changes. These effects were found to be associated with the suppression of the phosphoinositide 3‑kinase/AKT signaling pathway and the subsequent reduction of the expression of interleukin (IL)‑6, IL‑1β, and tumor necrosis factor‑α in the serum and myocardial tissue. These results indicate that BBR has the potential be an effective alternative therapy for the prevention and treatment of myocardial I/R injury in clinical practice.
Upregulates SIRT1
With a long history of application in Chinese traditional medicine, berberine (BBR) was reported to exhibit healthspan-extending properties in some age-related diseases, such as type 2 diabetes and atherosclerosis. However, the antiaging mechanism of BBR is not completely clear. By means of hydrogen peroxide- (H2O2-) induced premature cellular senescence model, we found that a low-concentration preconditioning of BBR could resist premature senescence in human diploid fibroblasts (HDFs) measured by senescence-associated β-galactosidase (SA-β-gal), accompanied by a decrease in loss of mitochondrial membrane potential and production of intracellular reactive oxygen species (ROS). Moreover, the low-concentration preconditioning of BBR could make cells less susceptible to subsequent H2O2-induced cell cycle arrest and growth inhibition. Experimental results further showed that the low concentration of BBR could induce a slight increase of ROS and upregulate the expression level of sirtuin 1 (SIRT1), an important longevity regulator.H2O2-induced activation of checkpoint kinase 2 (Chk2) was significantly attenuated after the preconditioning of BBR. The present findings implied that the low-concentration preconditioning of BBR could have a mitohormetic effect against cellular senescencetriggered by oxidative stress in some age-related diseases through the regulation of SIRT1.
MDM2 INHIBITION
MDM2 inhibition-mediated autophagy contributes to the pro-apoptotic effect of berberine in p53-null leukemic cells
Significance: MDM2 inhibits autophagy and apoptosis in leukemic cells in a p53-independent manner. BBR induces autophagy in p53-null leukemic cells through downregulating MDM2 expression at both transcriptional and post-transcriptional levels, which may contribute to the anti-cancer effect of BBR in leukemia.
EPHRIN-B2 & VEGFR2 INHIBITOR
Background: Berberine, a plant-derived compound isolated from Coptis chinensis used in traditional Chinese medicine, has been shown to possess anti-cancer properties. However, no study has shown that berberine could target ephrin-B2, which plays a critical role in cell proliferation and migration. Purpose: The aim of this study is to investigate the effect of berberine on cancer cell growth and migration, through the regulation of ephrin-B2 and downstream signaling molecules. Methods: In this study, a high ephrin-B2-expressing cell membrane chromatography method was developed to investigate 48 crude extracts from traditional Chinese medicine that could act on ephrin-B2. Cell proliferative and wound-healing assays were used to study the effect of berberine on cancer cell growth and migration. The mechanism of berberine was investigated using western blot. Results: Berberine was isolated from C. chinensis extracts and showed activity on the HEK293/ephrin-B2 cell membrane chromatography column. Berberine showed a greater inhibitory effect in high-expressing ephrin-B2 cells (HEK293/ephrin-B2 cells) than in normal HEK293 cells, and decreased the expression of ephrin-B2 and its PDZ binding proteins, which indicates that ephrin-B2 is a target of berberine.Furthermore, berberine downregulates the phosphorylation of VEGFR2and downstream signaling members (AKT and Erk1/2), which in turn downregulates the expression of MMP2 and MMP9. Conclusion: The above data confirm the inhibitory effects of berberine on ZR-75-30 cell proliferation and cell migration. Overall, our studies demonstrate that berberine inhibits cell growth and migration by targeting ephrin-B2.
In conclusion, results presented in this work demonstrate that berberine isolated from C. chinensis can interact with ephrin-B2, decrease the expression of ephrin-B2 and its PDZ binding proteins, and downregulate the phosphorylation of VEGFR2and downstream signaling members (AKT and Erk1/2), which in turn downregulates the expression of MMP2 and MMP9. All of these contribute to the inhibition of cell growth and cell migration by berberine. These data support the development of berberine as an ephrin-B2- mediated inhibitor of breast cancer ZR-75-30 cell growth.
EPHRIN-B2 IS ENHANCED IN SCAPS
Hypoxic conditions induced upregulated HIF-1α and VEGF expression in HUVECs, whereas ephrin-B2 gene was enhanced in SCAPs. Notably, this gene plays a central role in heart morphogenesis and angiogenesis by regulating cell adhesion and cell migration. Additionally, synergistic effects between HIF-1, VEGF, and ephrin-B2 led to an increase in the endothelial tubule number, vessel length, and branching points [92].
CDK1 INHIBITOR
Alkaloids, berberine and palmatine, isolated from the extracts of Berberis lycium Royle (Berberidacea) were found to inhibit proliferation of human promyelocytic HL-60 cells by the cell cycle arrest in S phase, as described in [130]. The compounds were shown to activate CHEK2, and degradation of CDC25A, and the subsequent inactivation of CDK1, as indicated in [130]. Berberine also downregulated the cyclin D1 expression, and induced the acetylation of α-tubulin [130].
Boswellic Acid
(from the fragrant gum resin of the Boswellia serrata tree)
Acetyl-11-keto-β-boswellic acid suppresses docetaxel-resistant prostate cancer cells in vitro and in vivo by blocking Akt and Stat3 signaling, thus suppressing chemoresistant stem cell-like properties
Acquired docetaxel-resistance of prostate cancer (PCa) remains a clinical obstacle due to the lack of effective therapies. Acetyl-11-keto-β-boswellic acid (AKBA) is a pentacyclic triterpenic acid isolated from the fragrant gum resin of the Boswellia serrata tree, which has shown intriguing antitumor activity against human cell lines established from PCa, colon cancer, malignant glioma, and leukemia. In this study, we examined the effects of AKBA against docetaxel-resistant PCa in vitro and in vivo as well as its anticancer mechanisms. We showed that AKBA dose-dependently inhibited cell proliferation and induced cell apoptosis in docetaxel-resistant PC3/Doc cells; its IC50 value in anti-proliferation was ∼17 μM. Furthermore, AKBA dose-dependently suppressed the chemoresistant stem cell-like properties of PC3/Doc cells, evidenced by significant decrease in the ability of mammosphere formation and down-regulated expression of a number of stemness-associated genes. The activation of Akt and Stat3 signaling pathways was remarkably enhanced in PC3/Doc cells, which contributed to their chemoresistant stem-like phenotype. AKBA (10–30 μM) dose-dependently suppressed the activation of Akt and Stat3 signaling pathways in PC3/Doc cells. In contrast, overexpression of Akt and Stat3 significantly attenuated the inhibition of AKBA on PC3/Doc cell proliferation. In docetaxel-resistant PCa homograft mice, treatment with AKBA significantly suppresses the growth of homograft RM-1/Doc, equivalent to its human PC3/Doc, but did not decrease their body weight. In summary, we demonstrate that AKBA inhibits the growth inhibition of docetaxel-resistant PCa cells in vitro and in vivo via blocking Akt and Stat3 signaling, thus suppressing their cancer stem cell-like properties.
Inhibition of Cyclooxygenase-2 and Mcl-1 Expression
Background: We have previously shown that berberine exerts its anti-inflammatory effects through inhibition of cyclooxygenase-2 (COX-2) expression. In this study, we explored the biochemical influence of berberine-induced COX-2 reduction and apoptosis. Materials and Methods: KB cells were treated with berberine, and the apoptosis was measured by morphology and caspase-3 activity. The effects of prostaglandin E2 (PGE2) on berberine-mediated cell growth were also determined. The expression of COX-2, Bcl-2, Mcl-1, Akt and phosphorylated Akt in berberine-treated KB cells, with or without PGE2, were assessed by Western blots. Results: Berberine induced apoptosis in KB cells, and was partially reversed by incorporation of PGE2. Berberine treatment inhibited COX-2 and Mcl-1 expression dose-dependently, but not Bcl-2. PGE2 induced COX-2 and Mcl-1 expression and reversed the repressive effect of berberine on Mcl-1. In addition, PGE2 had no effect on total Akt, but slightly reversed the phosphorylated Akt, which was decreased by berberine. Conclusion: These results suggest that berberine-induced apoptosis might be COX-2-dependent and is related to decreased Akt phosphorylation and Mcl-1 expression.
Betulinic acid
decreases expression of bcl-2, BCLw, MCL-1 and cyclin D1 / Increases BAX
Betulinic acid (BA) is a pentacyclic triterpene found in many plant species, among others in the bark of white birch Betula alba. BA was reported to display a wide range of biological effects, including antiviral, antiparasitic, antibacterial and anti-inflammatory activities, and in particular to inhibit growth of cancer cells. The aim of the study was further in vitro characterization of BA anticancer activity. In this study, we demonstrated a remarkable antiproliferative effect of BA in all tested tumor cell cultures including neuroblastoma, rabdomyosarcoma-medulloblastoma, glioma, thyroid, breast, lung and colon carcinoma, leukemia and multiple myeloma, as well as in primary cultures isolated from ovarian carcinoma, cervical carcinoma and glioblastoma multiforme. Furthermore, we have shown that BA decreased cancer cell motility and induced apoptotic cell death. We also observed decrease of bcl2 and cyclin D1genes expression, and increase of baxgene expression after betulinic acid treatment. These findings demonstrate the anticancer potential of betulinic acid and suggest that it may be taken into account as a supportive agent in the treatment of cancers with different tissue origin.

Brevilin A
(Sesquiterpene lactone from Centipeda Minima aka “Nakchhikni”)
BCL-XL Inhibitor
To get better insight into Brevilin A-induced apoptosis in U87 glioblastoma cells, we measured the expression of bcl-2 family proteins by Western blotting analysis. The data demonstrated that Brevilin A decreased the expression of anti-apoptotic Bcl-xL protein whereas it increased the expression of pro-apoptotic Bak protein. However, no change in the expression of anti-apoptotic Bcl-2 protein and pro-apoptotic Bax protein was observed. To support our data, we further measured the expression of cytochrome c in cytosolic fractions. The data showed that Brevilin A treatment induced release of cytochrome c from the mitochondria into the cytosol. Next, we determined mitochondrial membrane potential in U87 glioblastoma cells by using the JC-1 fluorescent probe. In healthy cells, JC-1 forms complexes known as J-aggregates, which show intense red fluorescence; however, in cells with disrupted MMP, JC-1 remains in monomeric form and manifests green fluorescence. Thus, MMP is calculated by a decrease in red/green fluorescence ratio. The data demonstrated that Brevilin A decreased red/green fluorescence ratio (MMP) in U87 glioblastoma cells in a dose-dependent manner. These findings clearly indicate that Brevilin A induces at least, in part, mitochondrial apoptosis in U87 glioblastoma cells.
potent Jak/stat inhibitor
Signal abnormalities in human cells usually cause unexpected consequences for individual health. We focus on these kinds of events involved in JAK-STAT signal pathways, especially the ones triggered by aberrant activated STAT3, an oncoprotein which participates in essential processes of cell survival, growth and proliferation in many types of tumors, as well as immune diseases. By establishing a STAT3 signal based high-throughput drug screening system in human lung cancer A549 cells, we have screened a library from natural products which contained purified compounds from medicinal herbs. One compound, named Brevilin A, exhibited both strong STAT3 signal inhibition and STAT3 signal dependent cell growth inhibition. Further investigations revealed that Brevilin A not only inhibits STAT3 signaling but also STAT1 signaling for cytokines induced phosphorylation of STAT3 and STAT1 as well as the expression of their target genes. In addition, we found Brevilin A could attenuate the JAKs activity by blocking the JAKs tyrosine kinase domain JH1. The levels of cytokine induced phosphorylation of STATs and other substrates were dramatically reduced by treatment of Brevilin A. The roles of Brevilin A targeting on JAKs activity indicate that Brevilin A may not only be used as a STAT3 inhibitor but also a compound blocking other JAK-STAT hyperactivation. Thus, these findings provided a strong impetus for the development of selective JAK-STAT inhibitors and therapeutic drugs in order to improve survival of patients with hyperactivated JAKs and STATs.
Inactivates PI3K/AKT/mTOR
Brevilin A is a sesquiterpene lactone isolated from Centipeda minima and possesses inhibitory effects on proliferation of various tumor cells. In this study, Brevilin A inhibitory effect on proliferation and its molecular mechanism of action were investigated both in vivo and in vitro in colon adenocarcinoma CT26 cells. The results indicated that the inhibitory effect of Brevilin A in CT26 proliferation was dose-dependent and this effect was due to apoptosis. Furthermore, Brevilin A increased ROS levels, decreased mitochondrial membrane potential (MMP) and induced apoptosis of CT26 cell in a dose-dependent manner. Apoptosis induced by Brevilin A was higher than that induced by adriamycin under the same dose. Cleaved-caspase-8, cleaved-caspase-9 and cleaved-caspase-3 were up-regulated after Brevilin A treatment, together with an increase of Bax protein expression, while Bcl-2 was reduced. Further investigation revealed that Brevilin A inhibited the phosphorylation of PI3K, AKT and mTOR and promoted the expressions of autophagy-related proteins LC3-II, Beclin1 and Atg5 and consequent formation of autophagosomes, whereas 3-methyladenine (3-MA), a type III PI3K inhibitor, inhibited autophagosomes formation induced by Brevilin A. In vivoinvestigation suggested that Brevilin A significantly inhibited the growth of CT26 tumor compared to adriamycin and concurrently promoted the expressions of LC3-II and cleaved-caspase-3 in tumor tissues. Our results demonstrated that the anti-tumor activity of Brevilin A was mainly achieved by the induction of cell apoptosis and autophagy, suggesting a promising potential as antitumor drug against colon adenocarcinoma.
Butein
Toxicodendron vernicifluum
Butein inhibits the growth of drug-resistant cancer cells (A2780cis and H1975) with modest potency. Butein induces a significant degradation of Hsp90 client proteins in A2780cis and H1975 cell lines. The biochemical and cellular studies demonstrates that butein inhibits the Hsp90 folding machinery. The result suggests that butein would be a potential therapeutic lead to overcome the drug resistance of cancer.
Taken together, these results indicate that butein could possibly sensitize to TRAIL-induced apoptosis by inhibiting the activation of NF-κB, including Rel-A. In contrast, recently important findings showed that the activation status of NF-κB is not sufficient to determine the fate of a cell with respect to TRAIL-induced apoptosis in hepatocellular carcinoma (10). Braeuer et al. (39) also reported that constitutively activated NF-κB, but not induced NF-κB, leads to TRAIL resistance by upregulation of XIAP in human cancer cells. Therefore, the effects of NF-κB will be investigated in TRAIL resistance. In addition, suppression of NF-κB activity by butein may also be involved in the stimulation of caspase-8 activity because the NF-κB–induced products, IAP-1, IAP-2, and XIAP, are known to cooperatively block caspase-8 activity (40).

Campesterol
down-regulates Bcl-2 and Bcl-xL / up-regulates Bax, Bad and Bak
This study demonstrates that Tb extract significantly inhibited A549 cell proliferation, while this extract affected the sensitivity of normal lung fibroblast cells to a lesser extent. Treating A549 cells with Tb extract led to a cell cycle arrest at the G2/M phase and induced apoptosis of A549 cells by down-regulating Bcl-2 and Bcl-xL protein expression and up-regulating Bax, Bad and Bak expression. Additionally, dibutyl phthalate, -linolenic acid, phytol, campesterol, stigmasterol and -sitosterol were the major effective bioactive compounds in Tb extract. Further studies are needed to fully elucidate the mechanisms involved in cancer cell death and to ensure that this extract is safe for human consumption.

Carnosic acid
down-regulation of c-FLIP and Bcl-2
Carnosic acid is a phenolic diterpene from rosmarinus officinalis, and has multiple functions, such as anti-inflammatory, anti-viral, and anti-tumor activity. In this study, we examined whether carnosic acid could sensitize TRAIL-mediated apoptosis in human renal carcinoma Caki cells. We found that carnosic acid markedly induced TRAIL-mediated apoptosis in human renal carcinoma (Caki, ACHN, and A498), and human hepatocellular carcinoma (SK-HEP-1), and human breast carcinoma (MDA-MB-231) cells, but not normal cells (TMCK-1 and HSF). Carnosic acid induced down-regulation of c-FLIP and Bcl-2 expression at the post-translational levels, and the over-expression of c-FLIP and Bcl-2 markedly blocked carnosic acid-induced TRAIL sensitization. Furthermore, carnosic acid induced death receptor (DR)5, Bcl-2 interacting mediator of cell death (Bim), and p53 up-regulated modulator of apoptosis (PUMA) expression at the transcriptional levels via CCAAT/enhancer-binding protein-homologous protein (CHOP). Down-regulation of CHOP expression by siRNA inhibited DR5, Bim, and PUMA expression, and attenuated carnosic acid plus TRAIL-induced apoptosis. Taken together, our study demonstrates that carnosic acid enhances sensitization against TRAIL-mediated apoptosis through the down-regulation of c-FLIP and Bcl-2expression, and up-regulation of ER stress-mediated DR5, Bim, and PUMA expression at the transcriptional levels.
CarNosine
(beta-alanyl-L-histidine)
DELAYS Cellular Senescence
Many diverse properties of the dipeptide carnosine, which are more completely described elsewhere in this volume, stimulated the idea that carnosine may have some useful therapeutic value, particularly with regard to old age. We were greatly inspired by the pioneering work of McFarland and Holliday [1], where it was shown and later confirmed [2] that the endogenous dipeptide carno- sine (β-alanyl-L-histidine) was able not only to rejuvenate human cells in cultures, but also influence the formation of long-lived clones affecting earlier events during serial subculture. The work of Kantha et al. who had also shown an anti-senescence effect of carnosine in vitro [3] encouraged us to test it on small mammals in order to obtain some in vivo data. The Senescence Accelerated Mice line (hereafter SAM) was chosen for our initial experiments because of the short lifespan of the animals and the already large amount of literature that exists about this line [4]. In SAM animals, an over-production of free radicals occurring in their tissues may cause the accelerated senescence [5-8]. The full details of our exper- iments are described elsewhere [9, 10]. These experiments revealed that carnosine at a daily dose of 100 mg/kg of body weight was able to extend the mean lifespan of the mice by 20% but had no effect on the maximum lifespan.The polypotent effects of carnosine which are described throughout this journal and the wider literature make it an ideal candidate as a so-called geropro- tectoran agent which may delay or prevent some conditions intrinsic with old age.
MTor Inhibitor (acts like rapamyacin mimetic)
Anti-ageing mechanisms of carnosine include inhibition of mTOR and TGFβ/Smad3 pathways, and suppressing the effects of reactive carbonyl compounds. The causes of ageing are usually regarded as multifactorial; thus effective regulation might be achieved by intervention at multiple sites. It has been suggested that the endogenous dipeptide carnosine, also available as a food supplement, possesses anti-ageing activity and may achieve its reported age-alleviating effects via a number of mechanisms. Carnosine’s possible anti-ageing mechanisms are therefore discussed; the evidence suggests that inhibition of the mechanistic target of rapamycin and carbonyl scavenging may be involved.
Carbonyl Scavenger
The metabolic syndrome is a risk factor that increases the risk for development of renal and vascular complications. This study addresses the effects of chronic administration of the endogenous dipeptide carnosine (β‐alanyl‐L‐histidine, L‐CAR) and of its enantiomer (β‐alanyl‐D‐histidine, D‐CAR) on hyperlipidaemia, hypertension, advanced glycation end products, advanced lipoxidation end products formation and development of nephropathy in the non‐diabetic, Zucker obese rat. The Zucker rats received a daily dose of L‐CAR or D‐CAR (30 mg/kg in drinking water) for 24 weeks. Systolic blood pressure was recorded monthly. At the end of the treatment, plasma levels of triglycerides, total cholesterol, glucose, insulin, creatinine and urinary levels of total protein, albumin and creatinine were measured. Several indices of oxidative/carbonyl stress were also measured in plasma, urine and renal tissue. We found that both L‐ and D‐CAR greatly reduced obese‐related diseases in obese Zucker rat, by significantly restraining the development of dyslipidaemia, hypertension and renal injury, as demonstrated by both urinary parameters and electron microscopy examinations of renal tissue. Because the protective effect elicited by L‐ and D‐CAR was almost superimposable, we conclude that the pharmacological action of L‐CAR is not due to a pro‐histaminic effect (D‐CAR is not a precursor of histidine, since it is stable to peptidic hydrolysis), and prompted us to propose that some of the biological effects can be mediated by a direct carbonyl quenching mechanism.
antiglycating Agent
During the process of glycation that happens with ageing, certain aldoses or aldehyde molecules attach to proteins (or to DNA) causing cross linking, that is, abnormal protein-to- protein or protein-to-DNA bonds. This process is facilitated by carbonyl groups. These abnormal proteins then may accumulate forming AGEs (Advance Glycation End-products) that may, in turn, react with free radicals to cause chronic degenerative diseases associated with aging. Apart from its antioxidant activities discussed above, carnosine has been found to possess significant anti-glycating properties, interfering with the glycation processes at several steps.
On the enigma of carnosine’s anti-ageing actions
Carnosine (�-alanyl-L-histidine) has described as a forgotten and enigmatic dipeptide. Carnosine’s enigma is particularly exemplified by its apparent anti-ageing actions; it suppresses cultured human fibroblast senescence and delays ageing in senescence-accelerated mice and Drosophila, but the mechanisms reponsible remain uncertain. In addition to carnosine’s well-documented anti-oxidant, anti-glycating, aldehyde-scavenging and toxic metal-ion chelating properties, its ability to influence the metabolism of altered polypeptides, whose accumulation characterises the senescent phenotype, should also be considered. When added to cultured cells, carnosine was found in a recent study to suppress phosphorylation of the translational initiation factor eIF4E resulting in decreased translation frequency of certain mRNA species. Mutations in the gene coding for eIF4E in nemtodes extend organism lifespan, hence carnosine’s anti-ageing effects may be a consequence of decreased error-protein synthesis which in turn lowers formation of protein carbonyls and increases protease availability for degradation of polypeptides altered postsynthetically. Other studies have revealed carnosine-induced upregulation of stress protein expression and nitric oxide synthesis, both of which may stimulate proteasomal elimination of altered proteins. Some anti-convulsants can enhance nematode longevity and suppress the effects of a protein repair defect in mice, and as carnosine exerts anti-convulsant effects in rodents, it is speculated that the dipeptide may participate in the repair of protein isoaspartyl groups. These new observations only add to the enigma of carnosine’s real in vivo functions. More experimentation is clearly required.
Caloric restriction mimetic
The long-term consumption of approximately 30–50% fewer calories than the amount consumed ad libitum (calorie restriction – CR), is perhaps the only widely accepted method of extending maximum lifespan in most animals studied so far. Not surprisingly, humans are not always willing to undergo a life-long CR diet, even if this may possibly mean an extension of the currently maximum human lifespan of around 110– 120 years. CR affects several genes, molecules, hormones, and other parameters, and during the past few years, there has been an attempt to identify compounds that may have biological activities similar to those of CR. Several such compounds have been identified and have been classified as Calorie Restriction Mimetics (CRM) [33]. It has been hypothesized [34] that CR may exert some of its benefits via suppression of glycolysis, thus reducing the formation of reactive oxygen species (ROS), and reducing the formation of glycating agents such as methyglyoxal (MG) [35]. As carnosine also reduces MG and ROS it may be considered as a CRM, mimicking several other physiological actions of CR itself [36].
Use of carnosine as a natural anti-senescence drug for human beings
Carnosine is an endogenous free-radical scavenger. The latest research has indicated that apart from the function of protecting cells from oxidation-induced stress damage, carnosine appears to be able to extend the lifespan of cultured cells, rejuvenate senescent cells, inhibit the toxic effects of amyloid peptide (A beta), malondialdehyde, and hypochlorite to cells, inhibit glycosylation of proteins and protein-DNA and protein-protein cross-linking, and maintain cellular homeostasis. Also, carnosine seems to delay the impairment of eyesight with aging, effectively preventing and treating senile cataract and other age-related diseases. Therefore, carnosine may be applied to human being as a drug against aging.
Carnosine, a protective, anti-ageing peptide
Carnosine (β-alanyl-l-histidine) has protective functions additional to anti-oxidant and free-radical scavenging roles. It extends cultured human fibroblast life-span, kills transformed cells, protects cells against aldehydes and an amyloid peptide fragment and inhibits, in vitro, protein glycation (formation of cross-links, carbonyl groups and AGEs) and DNA/protein cross-linking. Carnosine is an aldehyde scavenger, a likely lipofuscin (age pigment) precursor and possible modulator of diabetic complications, atherosclerosis and Alzheimer’s disease.
Effect of Carnosine on Age-Induced Changes in Senescence-Accelerated Mice
The effect of carnosine on the life span and several brain biochemical characteristics in senescence-accelerated mice—prone 1 (SAMP1) was investigated. A 50% survival rate of animals treated with carnosine increased by 20% as compared to controls. Moreover, the number of animals that lived to an old age significantly increased. The effect of carnosine on life span was accompanied by a decrease in the level of ′-tiobarbituric acid reactive substances (TBARS), monoamine oxidase b (MAO b), and Na/K-ATPase activity. There was also an increase in glutamate binding to N-methyl-D-aspartate receptors. These observations are consistent with the conclusion that carnosine increases life span and quality of life by diminishing production of lipid peroxides and reducing the influence of reactive oxygen species (ROS) on membrane proteins
Carnosine, the Protective, Anti-aging Peptide
Carnosine attenuates the development of senile features when used as a supplement to a standard diet of senescence accelerated mice (SAM). Its effect is apparent on physical and behavioral parameters and on average life span. Carnosine has a similar effect on mice of the control strain, but this is less pronounced due to the non-accelerated character of their senescence processes.
Reaction of Carnosine with Aged Proteins
Cellular aging is often associated with an increase in protein carbo- nyl groups arising from oxidation- and glycation-related phenomena and suppressed proteasome activity. These “aged” polypeptides may either be degraded by 20S proteasomes or cross-link to form structures intractable to proteolysis and inhibitory to proteasome activity. Carnosine is present at surprisingly high levels (up to 20 mM) in muscle and nervous tissues in many animals, especially long-lived species. Carnosine can delay senescence in cultured human fibroblasts and reverse the senescent phenotype, restoring a more juvenile appearance. As better antioxidants/free-radical scav- engers than carnosine do not demonstrate these antisenescent effects, addition- al properties of carnosine must contribute to its antisenescent activity. Having shown that carnosine can react with protein carbonyls, thereby generating “carnosinylated” polypeptides using model systems, we propose that similar adducts are generated in senescent cells exposed to carnosine. Polypeptide-car- nosine adducts have been recently detected in beef products that are relatively rich in carnosine, and carnosine’s reaction with carbonyl functions generated during amino acid deamidation has also been described. Growth of cultured human fibroblasts with carnosine stimulated proteolysis of long-labeled pro- teins as the cells approached their “Hayflick limit,” consistent with the idea that carnosine ameliorates the senescence-associated proteolytic decline. We also find that carnosine suppresses induction of heme-oxygenase-1 activity following exposure of human endothelial cells to a glycated protein. The antisenescent activity of the spin-trap agent -phenyl-N-t-butylnitrone (PBN) towards cultured human fibroblasts resides in N-t-butyl-hydroxylamine, its hydrolysis product. As hydroxylamines are reactive towards aldehydes and ketones, the antisenescent activity of N-t-butyl-hydroxylamine and other hydroxylamines may be mediated, at least in part, by reactivity towards macromolecular carbonyls, analogous to that proposed for carnosine.
Rejuvenates senescent cells
We have examined the effects of the naturally occurring dipeptide carnosine (β-alanyl-L-histidine) on the growth, morphology, and lifespan of cultured human diploid fibroblasts. With human foreskin cells, HFF-1, and fetal lung cells, MRC-5, we have shown that carnosine at high concentrations (20-50 mM) in standard medium retards senescence and rejuvenates senescent cultures. These late-passage cultures preserve a nonsenescent morphology in the presence of carnosine, in comparison to the senescent morphology first described by Hayflick and Moorhead. Transfer of these late-passage cells in medium containing carnosine to unsupplemented normal medium results in the appearance of the senescent phenotype. The serial subculture of cells in the presence of carnosine does not prevent the Hayflick limit to growth, although the lifespan in population doublings as well as chronological age is often increased. This effect is obscured by the normal variability of human fibroblast lifespans, which we have confirmed. Transfer of cells approaching senescence in normal medium to medium supplemented with carnosine rejuvenates the cells but the extension in lifespan is variable. Neither D -carnosine, (β-alanyl-D-histidine), homocarnosine, anserine, nor β-alanine had the same effects as carnosine on human fibroblasts. Carnosine is an antioxidant, but it is more likely that it preserves cellular integrity by its effects on protein metabolism.
JNK, Bcl-2↓, Bcl-xL↓, XIAP↓, caspase-3↑, caspase-9↑, cyclin B1↓, Bax↑, TNF↓, DR5↑, MMP2↓, MMP9↓, NF-κB↓, STAT3↓, FOXO3a↓, FoxM1↓, CDK1↓, cdc25B↓, cyclin B↓, p27KIP1↑, cyclin A↓, cFLIP↓, survivin↓, cytochrome c↑, Bid↑
We investigated the effect of casticin on apoptosis induced by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL). We found that casticin potentiated TRAIL-induced apoptosis in human colon cancer cells. Casticin downregulated cell survival proteins including Bcl-xL, Bcl-2, survivin,XIAP and cFLIP, and induced death receptor 5 (DR5), but had no effect on DR4 and decoy receptors (DcR1 or DcR2). Deletion of DR5 by siRNA significantly reduced the apoptosis induced by TRAIL and casticin. In addition, casticin induced reactive oxygen species (ROS) generation in a dose-dependent manner. Collectively, the present study showed that casticin potentiates TRAIL-induced apoptosis through downregulation of cell survival proteins and induction of DR5 mediated by ROS.

Celastrol
(from Tripterygium Wilfordii aka “Thunder God Vine”)
Anti-Senescence effects
Reactive oxygen species (ROS) production has been implicated in the promotion of cellular senescence. Celastrol, a quinone methide triterpenoid isolated from the Celastraceae family, exerts antioxidant effects and enhances autophagy in various cell types. Since autophagy serves an important role in regulating ROS, it was hypothesized that the antioxidant effect of celastrol is via enhanced autophagy, thus inhibiting cell senescence. Therefore, the present study used a Senescence β‑Galactosidase Staining kit, western blot analysis and cell cycle analysis to investigate whether celastrol alleviates angiotensin (Ang) II‑induced cellular senescence by upregulating autophagy in vascular smooth muscle cells (VSMCs). The results demonstrated that celastrol reduced Ang II‑induced senescence of VSMCs. Ang II‑induced generation of ROS and the subsequent VSMC senescence were counteracted by pretreatment with celastrol, determined by a ROS assay kit. Celastrol significantly upregulated VSMC autophagy, which reduced intracellular ROS and the subsequent cellular senescence induced by Ang II. Furthermore, celastrol markedly suppressed activity of the mechanistic target of rapamycin signaling pathway in VSMCs. In conclusion, the present study demonstrated that celastrol counteracts VSMC senescence probably by reducing ROS production via activation of autophagy, which may hold promise for the prevention and treatment of aging‑associated cardiovascular disorders such as atherosclerosis.
HSP90 Inhibitor
The molecular chaperone heat shock protein 90 (Hsp90) is required for the stabilization and conformational maturation of various oncogenic proteins in cancer. The loading of protein kinases to Hsp90 is actively mediated by the cochaperone Cdc37. The crucial role of the Hsp90-Cdc37 complex has made it an exciting target for cancer treatment. In this study, we characterize Hsp90 and Cdc37 interaction and drug disruption using a reconstituted protein system. The GST pull-down assay and ELISA assay show that Cdc37 binds to ADP-bound/nucleotide-free Hsp90 but not ATP-bound Hsp90. Celastrol disrupts Hsp90-Cdc37 complex formation, whereas the classical Hsp90 inhibitors (e.g. geldanamycin) have no effect. Celastrol inhibits Hsp90 ATPase activity without blocking ATP binding. Proteolytic fingerprinting indicates celastrol binds to Hsp90 C-terminal domain to protect it from trypsin digestion. These data suggest that celastrol may represent a new class of Hsp90 inhibitor by modifying Hsp90 C terminus to allosterically regulate its chaperone activity and disrupt Hsp90-Cdc37 complex.
NF-KB Inhibitor
The NF-κB pathway plays an important role in chronic inflammatory and autoimmune diseases. Recently, NF-κB has also been suggested as an important mechanism linking obesity, inflammation, and metabolic disorders. However, there is no current evidence regarding the mechanism of action of NF-κB inhibition in insulin resistance and diabetic nephropathy in type 2 diabetic animal models. We investigated the effects of the NF-κB inhibitor celastrol in db/db mice. The treatment with celastrol for 2 months significantly lowered fasting plasma glucose (FPG), HbA1C and homeostasis model assessment index (HOMA-IR) levels. Celastrol also exhibited significant decreases in body weight, kidney/body weight and adiposity. Celastrol reduced insulin resistance and lipid abnormalities and led to higher plasma adiponectin levels. Celastrol treatment also significantly mitigated lipid accumulation and oxidative stress in organs including the kidney, liver and adipose tissue. The treated group also exhibited significantly lower creatinine levels and urinary albumin excretion was markedly reduced. Celastrol treatment significantly lowered mesangial expansion and suppressed type IV collagen, PAI-1 and TGFβ1 expressions in renal tissues. Celastrol also improved abnormal lipid metabolism, oxidative stress and proinflammatory cytokine activity in the kidney. In cultured podocytes, celastrol treatment abolished saturated fatty acid-induced proinflammatory cytokine synthesis. Taken together, celastrol treatment not only improved insulin resistance, glycemic control and oxidative stress, but also improved renal functional and structural changes through both metabolic and anti-inflammatory effects in the kidney. These results suggest that targeted therapy for NF-κB may be a useful new therapeutic approach for the management of type II diabetes and diabetic nephropathy.
Celastrol induces apoptosis and autophagy via JNK ACTIVATION
Celastrol, a triterpene from traditional Chinese medicine, has been proved to possess potent anti-tumor effect on various cancers. However, the effect of celastrol on human osteosarcoma and the underlying mechanisms remains to be elucidated. We reported here that celastrol could inhibit cell proliferation by causing G2/M phase arrest. Exposure to celastrol resulted in the activation of caspase-3, -8, and -9, indicating that celastrol induced apoptosis through both extrinsic and intrinsic pathways. Autophagy occurred in celastrol-treated cells as evidenced by formation of autophagosome and accumulation of LC3B-II. The celastrol-induced cell death was remarkably restored by the combination of autophagy and apoptosis inhibitors. Furthermore, inhibition of apoptosis enhanced autophagy while suppression of autophagy diminished apoptosis. Celastrol also induced JNK activation and ROS generation. The JNK inhibitor significantly attenuated celastrol-triggered apoptosis and autophagy while ROS scavenger could completely reverse them. The ROS scavenger also prevented G2/M phase arrest and phosphorylation of JNK. Importantly, we found that celastrol had the similar effects on primary osteosarcoma cells. Finally, in vivo, celastrol suppressed tumor growth in the mouse xenograft model. Taken together, our results revealed that celastrol caused G2/M phase arrest, induced apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells. Celastrol is therefore a promising candidate for development of antitumor drugs targeting osteosarcoma.
Celastrol INHIBITS IAP1, IAP2, Bcl-2, Bcl-XL, c-FLIP, and survivin via NF-KB
Celastrol, a quinone methide triterpene derived from the medicinal plant Tripterygium wilfordii, has been used to treat chronic inflammatory and autoimmune diseases, but its mechanism is not well understood. Therefore, we investigated the effects of celastrol on cellular responses activated by TNF, a potent proinflammatory cytokine. Celastrol potentiated the apoptosis induced by TNF and chemotherapeutic agents and inhibited invasion, both regulated by NF-kappaB activation. We found that TNF induced the expression of gene products involved in antiapoptosis (IAP1, IAP2, Bcl-2, Bcl-XL, c-FLIP, and survivin), proliferation (cyclin D1 and COX-2), invasion (MMP-9), and angiogenesis (VEGF) and that celastrol treatment suppressed their expression. Because these gene products are regulated by NF-kappaB, we postulated that celastrol mediates its effects by modulating the NF-kappaB pathway. We found that celastrol suppressed both inducible and constitutive NF-kappaB activation. Celastrol was found to inhibit the TNF-induced activation of IkappaBalpha kinase, IkappaBalpha phosphorylation, IkappaBalpha degradation, p65 nuclear translocation and phosphorylation, and NF-kappaB-mediated reporter gene expression. Recent studies indicate that TNF-induced IKK activation requires activation of TAK1, and we indeed found that celastrol inhibited the TAK1-induced NF-kappaB activation. Overall, our results suggest that celastrol potentiates TNF-induced apoptosis and inhibits invasion through suppression of the NF-kappaB pathway.
Celastrol, a Triterpene, Enhances TRAIL-induced Apoptosis through the Down-regulation of Cell Survival Proteins and Up-regulation of Death Receptors*
Whether celastrol, a triterpene from traditional Chinese medicine, can modulate the anticancer effects of TRAIL, the cytokine that is currently in clinical trial, was investigated. As indicated by assays that measure plasma membrane integrity, phosphatidylserine exposure, mitochondrial activity, and activation of caspase-8, caspase-9, and caspase-3, celastrol potentiated the TRAIL-induced apoptosis in human breast cancer cells, and converted TRAIL-resistant cells to TRAIL-sensitive cells. When examined for its mechanism, we found that the triterpene down-regulated the expression of cell survival proteins including cFLIP, IAP-1, Bcl-2, Bcl-xL, survivin, and XIAP and up-regulated Bax expression. In addition, we found that celastrol induced the cell surface expression of both the TRAIL receptors DR4 and DR5. This increase in receptors was noted in a wide variety of cancer cells including breast, lung, colorectal, prostate, esophageal, and pancreatic cancer cells, and myeloid and leukemia cells. Gene silencing of the death receptor abolished the effect of celastrol on TRAIL-induced apoptosis. Induction of the death receptor by the triterpenoid was found to be p53-independent but required the induction of CAAT/enhancer-binding protein homologous protein (CHOP), inasmuch as gene silencing of CHOP abolished the induction of DR5 expression by celastrol and associated enhancement of TRAIL-induced apoptosis. We found that celastrol also induced reactive oxygen species (ROS) generation, and ROS sequestration inhibited celastrol-induced expression of CHOP and DR5, and consequent sensitization to TRAIL. Overall, our results demonstrate that celastrol can potentiate the apoptotic effects of TRAIL through down-regulation of cell survival proteins and up-regulation of death receptors via the ROS-mediated up-regulation of CHOP pathway.

CHICORIC ACID
CASPASE 3 ACTIVATOR, induces apoptosis in 3T3-L1 preadipocytes
Chicoric acid has been reported to possess various bioactivities. However, the antiobesity effects of chicoric acid remain poorly understood. In this study, we investigated the effects of chicoric acid on 3T3-L1 preadipocytes and its molecular mechanisms of apoptosis. Chicoric acid inhibited cell viability and induced apoptosis in 3T3-L1 preadipocytes which was characterized by chromatin condensation and poly ADP-ribose-polymerase (PARP) cleavage. Mitochondrial membrane potential (MMP) loss, Bax/Bcl-2 dysregulation, cytochrome c release, and caspase-3 activationwere observed, indicating mitochondria-dependent apoptosis induced by chicoric acid. Furthermore, PI3K/Akt and MAPK (p38 MAPK, JNK, and ERK1/2) signaling pathways were involved in chicoric acid-induced apoptosis. The employment of protein kinase inhibitors LY294002, SB203580, SP600125, and U0126 revealed that PI3K/Akt signaling pathway interplayed with MAPK signaling pathways. Moreover, chicoric acid induced reactive oxygen species (ROS) generation. Pretreatment with the antioxidant N-acetylcysteine (NAC) significantly blocked cell death and changes of Akt and MAPK signalings induced by chicoric acid. In addition, chicoric acid down regulated HO-1 and COX-2 via the PI3K/Akt pathway.
chlorogenic acid
strong matrix metalloproteinase-9 inhibitor
A phenolic compound responsible for anti-MMP-9, which is known to be involved in tumor cell invasion and metastasis, has been isolated from methanol extracts prepared from stem barks of Euonymus alatus by assay-guided fractionation. The compound has been identified as 5-caffeoylquinic acid (chlorogenic acid; CHA) by NMR and FAB-MS. CHA showed a strong inhibitory effect of matrix metalloproteinase (MMP)-9 activityin a concentration-dependent manner on zymography. The purified CHA inhibited MMP-9 activity with the IC50 of 30–50 nM. Furthermore, the cytotoxic survival curve showed that CHA does not have cytotoxic effects on cellular proliferation, when Hep3B cells were treated with various concentrations of CHA and cell viability was measured using the XTT assay. The present data suggest a clue for possible mechanisms of cancer chemoprevention by CHA and other naturally occurring phenolic compounds. The results also imply that useful cancer chemopreventive agents can be further identified by combinations of in vitro (as a first screen) and in vivo studies.
HMGB1 Inhibitor
Chlorogenic Acid Attenuates High Mobility Group Box 1 (HMGB1) Sepsis is a complex, multifactorial, rapidly progressive disease characterized by an overwhelming activation of the immune system and the countervailing antiinflammatory response. In the current study in murine peritoneal macrophages, chlorogenic acid suppressed endotoxin-induced high mobility group box 1 (HMGB1) release in a concentration-dependent manner. Administration of chlorogenic acid also attenuated systemic HMGB1 accumulation in vivo and prevented mortality induced by endotoxemia and polymicrobial sepsis. The mechanisms of action of chlorogenic acid included attenuation of the increase in toll-like receptor (TLR)-4 expression and suppression of sepsis-induced signaling pathways, such as c-Jun NH2-terminal kinase (JNK), p38 mitogenactivated protein kinase (MAPK) and nuclear factor (NF)-κB, which are critical for cytokine release. The protection conferred by chlorogenic acid was achieved through modulation of cytokine and chemokine release, suppression of immune cell apoptosis and augmentation of bacterial elimination. Chlorogenic acid warrants further evaluation as a potential therapeutic agent for the treatment of sepsis and other potentially fatal systemic inflammatory disorders.

Chlorophyll a (Ludwigia octovalvis)
activates the CD95 (APO-1/CD95) system and AMPK pathway in 3T3-L1 cells
Ludwigia octovalvis is an aquatic plant widely distributed in Taiwan. It is traditionally used as a diuretic and is consumed as health drink. In this study, we evaluated the anti-proliferative activity of extracts and active constituent (chlorophyll a; CHL-a) of L. octovalvis in 3T3-L1 adipocytes; its mode of action on apoptosis was also investigated. Results showed that, among the different extracts and fractions, the ethylacetate layer (EAL) possessed the most potent anti-proliferative activity. Activity guided fractionation of the EAL obtained the bioactive constituent CHL-a(IC50: 24.10 ± 0.83 nM). At concentrations 5–30 nM, CHL-a exhibited a dose-dependent accumulation of the Sub-G1 peak and caused cell cycle arrest at the G0/G1 phase. At 30 nM, it significantly reduced the cell viability, induced the appearance of DNA fragments, and enhanced the activation of caspase-3. Western blot data revealed that CHL-a decreased the level of Bcl-2, and increased the expression of CD95 (APO-1/CD95) and Bax. Furthermore, CHL-a up-regulated the AMPK and p-AMPK levels, and down-regulated the expression of PPAR-γ. These results conclude that CHL-a possesses potent anti-proliferative activity, and its apoptotic effects on 3T3-L1 adipocytes are mediated through the activation of CD95 (APO-1/CD95) system and the AMPK signaling pathway.

Bcl-2↓, caspase-3↑, -8↑, and -9↑, Bax↑, Fas↑, Cdc2↓, cyclin B1↓; p21WAF1↑, procaspase-8↑, procaspase-3↑; JNK↑; PI3-K; PKC; ERK↑, NF-κB↓, cyclin E↓; p21↑, VEGF↓

Curcumin
(from Curcuma longa aka Turmeric Root)
Senolytic activity
Curcumin and o-Vanillin cleared senescent intervertebral disc (IVD) cells and reduced the senescence-associated secretory phenotype (SASP) associated with inflammation and back pain. Cells from degenerate and non-mildly-degenerate human IVD were obtained from organ donors and from patients undergoing surgery for low back pain. Gene expression of senescence and SASP markers was evaluated by RT-qPCR in isolated cells, and protein expression of senescence, proliferation, and apoptotic markers was evaluated by immunocytochemistry (ICC). The expression levels of SASP factors were evaluated by enzyme-linked immunosorbent assay (ELISA). Matrix synthesis was verified with safranin-O staining and the Dimethyl-Methylene Blue Assay for proteoglycan content. Western blotting and ICC were used to determine the molecular pathways targeted by the drugs. We found a 40% higher level of senescent cells in degenerate compared to non-mildly-degenerate discs from unrelated individuals and a 10% higher level in degenerate compared to non-mildly-degenerate discs from the same individual. Higher levels of senescence were associated with increased SASP. Both drugs cleared senescent cells, and treatment increased the number of proliferating as well as apoptotic cells in cultures from degenerate IVDs. The expression of SASP factors was decreased, and matrix synthesis increased following treatment. These effects were mediated through the Nrf2 and NFkB pathways.
Protective effect of curcumin against d-galactose-induced senescence in mice
Brain senescence plays an important role in cognitive dysfunction and neurodegenerative disorders. Curcumin was reported to have beneficial effect against several neurodegenerative disorders including Alzheimer’s disease. Therefore, the present study was conducted in order to explore the possible role of curcumin against d-galactose-induced cognitive dysfunction, oxidative damage, and mitochondrial dysfunction in mice. Chronic administration of d-galactose for 6 weeks significantly impaired cognitive function (both in Morris water maze and elevated plus maze), locomotor activity, oxidative defense (raised lipid peroxidation, nitrite concentration, depletion of reduced glutathione and catalase activity), and mitochondrial enzyme complex activities (I, II, and III) as compared to vehicle treated group. Curcumin (15 and 30 mg/kg) and galantamine (5 mg/kg) treatment for 6 weeks significantly improved cognitive tasks, locomotor activity, oxidative defense, and restored mitochondrial enzyme complex activity as compared to control (d-galactose). Chronic d-galactose treatment also significantly increased acetylcholine esterase activity that was attenuated by curcumin (15 and 30 mg/kg) and galantamine (5 mg/kg) treatment. In conclusion, the present study highlights the therapeutic potential of curcumin against d-galactose induced senescence in mice.
Curcumin prevents mitochondrial dysfunction in brain of senescence-accelerated mouse
The aging brain suffers mitochondrial dysfunction and a reduced availability of energy in the form of ATP, which in turn may cause or promote the decline in cognitive, sensory, and motor function observed with advancing age. There is a need for animal models that display some of the pathological features of human brain aging in order to study their prevention by e.g. dietary factors. We thus investigated the suitability of the fast-aging senescence-accelerated mouse-prone 8 (SAMP8) strain and its normally aging control senescence-accelerated mouse-resistant 1 (SAMR1) as a model for the age-dependent changes in mitochondrial function in the brain. To this end, 2-months old male SAMR1 (n = 10) and SAMP8 mice (n = 7) were fed a Western type diet (control groups) for 5 months and one group of SAMP8 mice (n = 6) was fed an identical diet fortified with 500 mg curcumin per kg. Dissociated brain cells and brain tissue homogenates were analyzed for malondialdehyde, heme oxygenase-1 mRNA, mitochondrial membrane potential (MMP), ATP concentrations, protein levels of mitochondrial marker proteins for mitochondrial membranes (TIMM, TOMM), the mitochondrial permeability transition pore (ANT1, VDAC1, TSPO), respiration complexes, and fission and fusion (Fis, Opa1, Mfn1, Drp1). Dissociated brain cells isolated from SAMP8 mice showed significantly reduced MMP and ATP levels, probably due to significantly diminished complex V protein expression, and increased expression of TSPO. Fission and fusion marker proteins indicate enhanced mitochondrial fission in brains of SAMP8 mice. Treatment of SAMP8 mice with curcumin improved MMP and ATP and restored mitochondrial fusion, probably by up-regulating nuclear factor PGC1α protein expression. In conclusion, SAMP8 compared to SAMR1 mice are a suitable model to study age-dependent changes in mitochondrial function and curcumin emerges as a promising nutraceutical for the prevention of neurodegenerative diseases that are accompanied or caused by mitochondrial dysfunction. Highlights ► Mitochondrial dysfunction and reduced energy production occur in the aging brain. ► We studied if SAMP8 vs. SAMR1 mice can be used as a model for mitochondrial function. ► Mitochondrial function was reduced and fission enhanced in SAMP8 vs. SAMR1 mice. ► Curcumin-intake restored mitochondrial function & fusion, perhaps by activating PGC1α. ► Age-dependent changes in brain mitochondrial function can be prevented by curcumin.
The Role of Curcumin in the Modulation of Ageing
It is believed that postponing aging is more effective and less expensive than the treatment of particular age-related diseases. Compounds which could delay symptoms of ageing, especially natural products present in a daily diet, are intensively studied. One of them is curcumin. It causes the elongation of the lifespan of model organisms, alleviates ageing symptoms and postpones the progression of age-related diseases in which cellular senescence is directly involved. It has been demonstrated that the elimination of senescent cells significantly improves the quality of life of mice. There is a continuous search for compounds, named senolytic drugs, that selectively eliminate senescent cells from organisms. In this paper, we endeavor to review the current knowledge about the anti-aging role of curcumin and discuss its senolytic potential.
Although treatment of Hodgkin’s lymphoma (HL) with a multi‐drug approach has been very successful, its toxicity becomes evident after several years as secondary malignancies and cardiovascular disease. Therefore, the current goal in HL treatment is to find new therapies that specifically target the deregulated signaling cascades, such as NF‐κB and STAT3, which cause Hodgkin and Reed‐Sternberg (H‐RS) cell proliferation and resistance of apoptosis. Based on the above information, we investigated the capacity of curcumin to inhibit NF‐κB and STAT3 in H‐RS cells, characterizing the functional consequences. Curcumin is incorporated into H‐RS cells and acts inhibiting both NF‐κB and STAT3 activation, leading to a decreased expression of proteins involved in cell proliferation and apoptosis,e.g. Bcl‐2, Bcl‐xL, cFLIP, XIAP, c‐IAP1, survivin, c‐myc and cyclin D1. Interestingly, curcumin caused cell cycle arrest in G2‐M and a significant reduction (80–97%) in H‐RS cell viability. Furthermore, curcumin triggered cell death by apoptosis, as evidenced by the activation of caspase‐3 and caspase‐9, changes in nuclear morphology and phosphatidylserine translocation. The above findings provide a mechanistic rationale for the potential use of curcumin as a therapeutic agent for patients with HL.
INHIBITS Bcl-2 & BCL-XL
Curcumin, a natural, biologically active compound extracted from rhizomes of Curcuma species, has been shown to possess potent anti-inflammatory, anti-tumor and anti-oxidative properties. The mechanism by which curcumin initiates apoptosis remains poorly understood. In the present report we investigated the effect of curcumin on the activation of the apoptotic pathway in human renal Caki cells. Treatment of Caki cells with 50 microM curcumin resulted in the activation of caspase 3, cleavage of phospholipase C-gamma1 and DNA fragmentation. Curcumin-induced apoptosis is mediated through the activation of caspase, which is specifically inhibited by the caspase inhibitor, benzyloxycarbony-Val-Ala-Asp-fluoromethyl ketone. Curcumin causes dose-dependent apoptosis and DNA fragmentation of Caki cells, which is preceded by the sequential dephosphorylation of Akt, down-regulation of the anti-apoptotic Bcl-2, Bcl-XL and IAP proteins, release of cytochrome c and activation of caspase 3. Cyclosporin A, as well as caspase inhibitor, specifically inhibit curcumin-induced apoptosis in Caki cells. Pre-treatment with N-acetyl-cysteine, markedly prevented dephosphorylation of Akt, and cytochrome c release, and cell death, suggesting a role for reactive oxygen species in this process. The data indicate that curcumin can cause cell damage by inactivating the Akt-related cell survival pathway and release of cytochrome c, providing a new mechanism for curcumin-induced cytotoxicity.
Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin’s lymphoma cells.
Although treatment of Hodgkin’s lymphoma (HL) with a multi-drug approach has been very successful, its toxicity becomes evident after several years as secondary malignancies and cardiovascular disease. Therefore, the current goal in HL treatment is to find new therapies that specifically target the deregulated signaling cascades, such as NF-kappaB and STAT3, which cause Hodgkin and Reed-Sternberg (H-RS) cell proliferation and resistance of apoptosis. Based on the above information, we investigated the capacity of curcumin to inhibit NF-kappaB and STAT3 in H-RS cells, characterizing the functional consequences. Curcumin is incorporated into H-RS cells and acts inhibiting both NF-kappaB and STAT3 activation, leading to a decreased expression of proteins involved in cell proliferation and apoptosis, e.g. Bcl-2, Bcl-xL, cFLIP, XIAP, c-IAP1, survivin, c-myc and cyclin D1. Interestingly, curcumin caused cell cycle arrest in G2-M and a significant reduction (80-97%) in H-RS cell viability. Furthermore, curcumin triggered cell death by apoptosis, as evidenced by the activation of caspase-3 and caspase-9, changes in nuclear morphology and phosphatidylserine translocation. The above findings provide a mechanistic rationale for the potential use of curcumin as a therapeutic agent for patients with HL.
Curcumin, a Dietary Component, Has Anticancer, Chemosensitization, and Radiosensitization Effects by Down-regulating the MDM2Oncogene through the PI3K/mTOR/ETS2 Pathway
The oncoprotein MDM2, a major ubiquitin E3 ligase of tumor suppressor p53, has been suggested as a novel target for human cancer therapy based on its p53-dependent and p53- independent activities. We have identified curcumin, which has previously been shown to have anticancer activity, as an inhibitor of MDM2 expression.
Cucurbitacin D
Disruptor of the HSP90 Chaperone Machinery
Heat shock protein 90 (Hsp90) facilitates the maturation of many newly synthesized and unfolded proteins (clients) via the Hsp90 chaperone cycle, in which Hsp90 forms a heteroprotein complex and relies upon cochaperones, immunophilins, etc., for assistance in client folding. Hsp90 inhibition has emerged as a strategy for anticancer therapies due to the involvement of clients in many oncogenic pathways. Inhibition of chaperone function results in client ubiquitinylation and degradation via the proteasome, ultimately leading to tumor digression. Small molecule inhibitors perturb ATPase activity at the N-terminus and include derivatives of the natural product geldanamycin. However, N-terminal inhibition also leads to induction of the pro-survival heat shock response (HSR), in which displacement of the Hsp90-bound transcription factor, heat shock factor-1, translocates to the nucleus and induces transcription of heat shock proteins, including Hsp90. An alternative strategy for Hsp90 inhibition is disruption of the Hsp90 heteroprotein complex. Disruption of the Hsp90 heteroprotein complex is an effective strategy to prevent client maturation without induction of the HSR. Cucurbitacin D, isolated from Cucurbita texana, and 3-epi-isocucurbitacin D prevented client maturation without induction of the HSR. Cucurbitacin D also disrupted interactions between Hsp90 and two cochaperones, Cdc37 and p23.
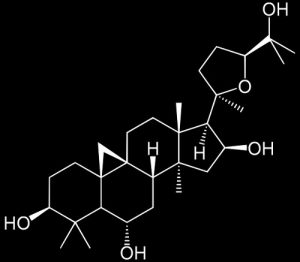
Cycloastragenol
STAT3 INHIBITOR
Cycloastragenol can negate constitutive STAT3 activation and promote paclitaxel-induced apoptosis in human gastric cancer cells
We observed that CAG exhibited cytotoxic activity against SNU-1 and SNU-16 cells to a greater extent as compared to normal GES-1 cells. CAG predominantly caused negative regulation of STAT3 phosphorylation at tyrosine 705 through the abrogation of Src and Janus-activated kinases (JAK1/2) activation. We noted that CAG impaired translocation of STAT3protein as well as its DNA binding activity. It further decreased cellular proliferation and mediated its anticancer effects predominantly by causing substantial apoptosis rather than autophagy. In addition, CAG potentiated paclitaxel-induced anti-oncogenic effects in gastric tumor cells.
Conclusions: Our results indicate that CAG can function to impede STAT3 activation in human gastric tumor cells and therefore it may be a suitable candidate agent for therapy of gastric cancer.
Telomerase activation and lengthening of telomeres
Cycloastragenol (CAG) is the aglycone derivative of astragaloside IV which has recently been demonstrated to activate telomerase and represents a potential drug candidate for the treatment of degenerative diseases. In the present study, intestinal absorption and metabolism of CAG were examined using the Caco-2 model and liver microsomes, respectively. The results showed that CAG could rapidly pass through the Caco-2 cell monolayer by passive diffusion. Four different glucuronide conjugates and two oxidized CAG metabolites were found in the apical and basolateral side of Caco-2 monolayer, suggesting that first-pass intestinal metabolism of CAG might occur upon passage through the intestinal epithelium. CAG underwent extensive metabolism in rat and human liver microsomes with only 17.4% and 8.2%, respectively, of the starting amount of CAG remaining after 30 min of incubation. Monohydroxylation of the parent and oxidization of the hydroxylated CAG were found in the liver samples. The present study indicated that CAG can be efficiently absorbed through intestinal epithelium. However, extensive first-pass hepatic metabolism would limit the oral bioavailability of this compound.
ANTI-AGING EFFECTS
Cycloastragenol (CAG) is a triterpenoid saponin compound and a hydrolysis product of the main active ingredient in Astragalus membranaceus (Fisch.) Bunge. An increasing body of evidence has indicated that CAG has a wide spectrum of pharmacological functions, which are attracting attention in the research community. The aim of the present review paper was to review and elucidate the advanced study of CAG. The focus was on advanced studies of CAG in English and Chinese databases; the literature was collected and reviewed to summarize the latest efficacy, pharmacokinetics and adverse reactions of CAG. Extensive pharmacological effects have been attributed to CAG, including telomerase activation, telomere elongation, anti‑inflammatory and anti‑oxidative properties; CAG has also been reported to improve lipid metabolism. Clinical research has demonstrated that CAG activates telomerase in humans and ameliorates various biomarkers. CAG is absorbed through the intestinal epithelium via passive diffusion and undergoes first‑pass hepatic metabolism. Within a certain dose range, oral CAG is relatively safe; however, underlying mechanisms associated with CAG are not clear, and thus, we should be aware of potential adverse reactions associated with CAG. According to existing studies and clinical trials, CAG is safe and has broad application prospects.
daidzein
BCL-2/BAX INHIBITOR
Daidzein induces apoptosis via the extrinsic receptor-mediated pathway, intrinsic mitochondrial pathway or endoplasmic reticulum stress pathway, depending on the type of tumor (21). For example, daidzein induces tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-mediated apoptosis in prostate cancer cells (41), however activates the mitochondria-mediated pathway in breast cancer, gastric carcinoma and hepatic cancer (22–24). Vilela et al (42) reported that bio-transformed soybean extract containing daidzein increases expression of TRAIL and its receptor DR4 in melanoma, resulting in cell apoptosis. Equol, which is a metabolite of daidzein, induces apoptosis in SMMC-7721 human hepatocellular carcinoma cells through the intrinsic and endoplasmic reticulum stress pathways (26). Caspase-9 and the Bcl-2/Bax ratio are commonly used activity markers of the mitochondrial apoptotic pathway (43–45). In the present study, western blotting was used to detect these markers, revealing that the cleavage of caspase-9 increased and the ratio of Bcl-2/Bax decreased in both cell lines in a dose-dependent manner, which indicated that daidzein-induced apoptosis was mediated via the mitochondrial apoptotic pathway. This process is similar to that in the human gastric carcinoma cell line BGC-823, as Tang et al (23) demonstrated that daidzein induces apoptosis via downregulation of Bcl-2/Bax and triggering of the mitochondrial pathway.
In the present study, the in vitro cytotoxicity of daidzein was evaluated in human BEL-7402, A549, HeLa, HepG-2 and MG-63 cancer cell lines. BEL-7402 cells were sensitive to daidzein treatment, with an IC50 value of 59.7±8.1 µM. Daidzein showed no cytotoxic activity toward A549, HeLa, HepG-2 and MG-63 cells. Daidzein increased the levels of reactive oxygen species (ROS) and induced a decrease in mitochondrial membrane potential. Morphological and comet assays showed that daidzein effectively induced apoptosis in BEL-7402 cells. Additionally, daidzein caused cell cycle arrest at the G2/M phase in the BEL-7402 cell line. Daidzein downregulated the expression of Bcl-2, Bcl-x and Baid proteins and upregulated the levels of Bim protein in the BEL-7402 cells. The results demonstrated that daidzein induced BEL-7402 cell apoptosis through an ROS-mediated mitochondrial dysfunction pathway.
Decursin
Downregulates STAT3, BCL-2, BCL-XL, SURVIVIN & VEGF
Recent reports have indicated that decursin can induce apoptosis, suppress tumor growth, and inhibit angiogenesis. In this experiment, we investigated how decursin could potentiate the cytotoxic effects of bortezomib in human multiple myeloma cells. We found that decursin inhibited cell viability in U266, MM.1S and ARH77 cells, but not in peripheral blood mononuclear cells (PBMC). Decursin-induced apoptosis through the activation of caspase-8, -9, and -3 in U266 cells. This correlated with the down-regulating of cyclin D1, bcl-2, bcl-xL, survivin, and the vascular endothelial growth factor (VEGF), which are all regulated by the activation of signal transducers and the activator of transcription 3 (STAT3). Indeed, decursin inhibited constitutive STAT3 activation through inhibition of the activation of Janus-activated kinase 2 (JAK2) in U266 cells. In addition, decursin inhibited interleukin-6-inducible STAT3 activation in a time-dependent manner in MM.1S cells. Interestingly, decursin significantly potentiated the apoptotic effects of bortezomib in U266 cells. These effects of decursin were correlated with the suppression of constitutive STAT3 activation in U266 cells. Overall, these results suggest that decursin is a novel blocker of STAT3 activation and it may be a potential candidate for overcoming chemo-resistance through suppression of this signaling.
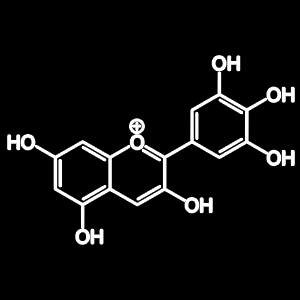
Delphinidin
Downregulates BCL-2 and BCL-XL
Solar UV radiation, in particular its UVB component, is the primary cause of many adverse biological effects, the most damaging of which is skin cancer. Here, we assessed the photochemopreventive effect of delphinidin, a major anthocyanidin present in many pigmented fruits and vegetables, on UVB-mediated responses in human immortalized HaCaT keratinocytes and SKH-1 hairless mouse skin. We found that pretreatment of cells with delphinidin (1-20 microM for 24 hours) protected against UVB (15-30 mJ/cm2, 24 hours)-mediated (i) decrease in cell viability and (ii) induction of apoptosis. Furthermore, we found that pretreatment of HaCaT cells with delphinidin inhibited UVB-mediated (i) increase in lipid peroxidation; (ii) formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG); (iii) decrease in proliferating cell nuclear antigen expression; (iv) increase in poly(ADP-ribose) polymerase cleavage; (v) activation of caspases; (vi) increase in Bax;(vii) decrease in Bcl-2; (viii) upregulation of Bid and Bak; and (ix) downregulation of Bcl-xL. Topical application of delphinidin (1 mg/0.1 ml DMSO/mouse) to SKH-1 hairless mouse skin inhibited UVB-mediated apoptosis and markers of DNA damage such as cyclobutane pyrimidine dimers and 8-OHdG. Taken together our results suggest that treatment of HaCaT cells and mouse skin with delphinidin inhibited UVB-mediated oxidative stress and reduced DNA damage, thereby protecting the cells from UVB-induced apoptosis.
Induces ROS accumulation; suppresses EGFR; damages DNA; downregulates HIF-1, p27, PI3K/Akt/mTOR, and NFƙB; induces caspase-3, -8, and -9
Delphinidin is one of the main compounds in blueberries, and an anticancer effect of delphinidin on a human colon cancer cell line (colo205) has been reported 126. The inhibitory effect of delphinidin on LoVo/ADR cell lines was also investigated. The data revealed that delphinidin inhibited metastatic CRC and that this may have been due to cellular ROS accumulation 125. Further investigation of delphinidin indicated that it inhibited HT-29 human tumor cells through the suppression of EGFR 97. A recent study on delphinidin claimed that it had antioxidant activity against human CRC HTC-116 and HT-29 cells and could also induce DNA damage 127. In addition, delphinidin potently inhibited HTC-116 and HT-29 cell lines through the downregulation of HIF-1 and p27 by affecting the PI3K/Akt/mTOR signaling pathway 128. HCT-116 cells treated with delphinidin suppressed the NF-kappa B pathway and activated the expression of caspase-3, -8 and -9, resulting in cell cycle arrest in the G2/M phase, thereby leading to apoptosis 129.
Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity
SENOLYTIC EFFECT
Digoxin is senolyticin lung fibrosis. Next, we wanted to test the senolytic effect of Digoxin using another in vivo model of a non- tumor related disease. For this, we established a mouse model of lung fibrosis induced by intratracheal administration of senescent human cells. Normal proliferating or gamma-irradiated (g-IR) senescent human fibroblasts IMR90 were delivered into the lungs of immunodeficient mice. Cell death analysis of in vitro treatment with Digoxin of proliferating and g-IR IMR90 confirmed once again the senolytic effect of the CG . Three weeks after intratracheal instillation, these animals were subjected to Digoxin or vehicle treatment for ten days and their lungs were removed and analyzed. First, we measured the expression levels of CDKN2A (coding for p16INK4a) a typical marker of primary fibroblast senescence42. Lungs from animals injected with g-IR IMR90 showed high levels of this human gene but not of the mouse ortholog, Cdkn2a, indicative of the presence of senescent human cells in the mouse lungs . Similarly, human CDKN1A (coding for p21) was also detected in the lungs of mice receiving senescent human cells . Treatment with Digoxin caused a significant reduction in CDKN2A expression levels and of CDKN1A (although not reaching statistical significance for this gene), suggesting that Digoxin caused the clearance of the human senescent cells. Finally, we stained the lungs with Masson Trichrome, a well-established marker of fibrosis. Lungs from animals injected with senescent g-IR IMR90 and treated with vehicle stained positive, indicative of fibrosis, while those treated with Digoxin had a reduced score of fibrosis. In addition, we determined hydroxyproline content in lungs as a measure of fibrosis and found that animals receiving senescent g-IR IMR90 cells had a significantly higher level of this marker compared to the ones injected with proliferating cells (Supple- mentary Fig. 5b). In line with our results using Masson Tri- chrome staining, mice treated with Digoxin showed a tendency to have reduced levels of hydroxyproline.
These results show the potential senolytic effect exerted by Digoxin in vivo using a model of a disease, other than cancer, caused by the accumulation of senescent cells.

Diosmin
Downregulates BCL-2
Reperfusion of ischemic tissue leads to the generation of oxygen derived free radicals which plays an important role in cellular damage. Objective of the current study is to evaluate the cardio-protective and antioxidant effect of diosmin on ischemia–reperfusion related cardiac dysfunction, oxidative stress and apoptosis. Diosmin (50 and 100 mg/kg body weight (bw)) was given every day to the rats orally throughout the experimental period. Ischemia/reperfusion protocol was carried out ex vivo using langendorff perfusion method and the cardiac functional recovery was assessed in terms of percentage rate pressure product. Coronary effluents of LDH and CK-MB activities, antioxidant enzyme activities, lipid peroxidation products, activity of TCA cycle enzymes were evaluated. Moreover, in vitro superoxide anionand hydroxyl radical scavenging potential of diosmin was also quantified. Finally, quantitative real-time PCRwas used for assessing Bcl-2 mRNA expression in heart. Cardiac functional recovery was impaired after reperfusion compared with continuously perfused heart. It was significantly prevented by diosmin treatment. Impaired antioxidant enzyme activities and elevated lipid peroxidation products level were also significantly suppressed. The activity of TCA cycle enzymes was protected against reperfusion stress. Down regulated Bcl-2 was also significantly increased.This study concluded that diosmin pretreatment prevents all the impaired patterns including cardiac function, oxidative stress and apoptosis associated with reperfusion in control heart by its antioxidant role.
IL-6 / STAT3 Inhibitor
Diosmin is naturally found flavanoid in many citrus fruits known to have anti-inflammatory, antihyperglycemic, antioxidant and antimutagenic properties. Effects of Diosmin on IL-6/STAT-3 expression in hamster buccal pouch carcinogenesisremain unclear. Alterations in many genes encode crucial proteins, which regulate cell proliferation, differentiation and apoptosis have been implicated in oral cancer. In the present study, we investigated the effect of dietary Diosmin on IL-6/STAT-3 signaling in the 7,12-dimethylbenz[a]anthracene (DMBA)-induced hamster buccal pouch (HBP) carcinogenesis by examining the protein expression of IL-6/STAT-3 and its related genes. Immunoblotting and immunohistochemical analyses revealed that Diosmin (100 mg/kg b.w) supplement inhibits key events in signaling especially STAT-3 phosphorylation and subsequent nuclear translocation. Results revealed that inhibition of proliferation and angiogenesis is associated with regulation of the STAT-3 pathway; where Diosmin prevents phosphorylation of JAK-1 which was ascend by IL-6, thereby inhibiting STAT-3 phosphorylation. Consequently, an imbalance in the Bax/Bcl-2 ratio triggered the caspase cascade in favor of apoptosis. Transmission electron microscopic studies proved the effect of Diosmin on ultrastructural changes. Finally our results provide significant evidence that Diosmin prevents the development and progression of HBP carcinomas through the inhibition of IL-6/STAT-3 signaling and its downstream events. Thus, Diosmin functions as a potent inhibitor of tumor development and progression by targeting IL-6/STAT-3 signaling may be an ideal candidate for cancer chemoprevention.

DX-9386
(a traditional Chinese formula: ginseng extract, acorus extract, polygala extract and hoelen extract)
Anti-aging effect of DX-9386 in senescence accelerated mouse.
The effects of DX-9386, a traditional Chinese prescription (ginseng, acorus, polygala and hoelen) were studied on life span, the degree of senescence, motor activity and the antibody production response in senescence accelerated mouse (SAM). DX-9386-containing food was given to SAM for 13 consecutive months from 2 months of age. DX-9386 significantly prolonged the life spanof SAM, prevented body weight decrease with aging and tended to improve the senile syndrome. The motor activity of SAMP8 was higher than that of SAMR1, and DX-9386 tended to increase the activity in SAMP8. The in vivo antibody production was markedly decreased in SAMP8 and DX-9386 showed no ameliorating effect on that. These results suggest that DX-9386 has anti-aging impact.
Ameliorative effects of chronic treatment using DX-9386, a traditional Chinese prescription, on learning performance and lipid peroxide content in senescence accelerated mouse.
DX-9386 is a traditional Chinese prescription consisting of ginseng, polygala, acorus and hoelen. The effect of chronic oral treatment with this preparation on learning behaviors and lipid peroxide concentration was studied in senescence accelerated mouse (SAM). SAM P8, a senescence prone of SAM, and SAM R1, a senescence resistant substrain of SAM, were started on a diet containing 1% of DX-9386 from the age of 2 months. All the experiments were performed at the age of 10 months. The prescription ameliorated the memory disorders of SAM P8, as evaluated in a step down test as well as a spatial memory test. The preparation, however, did not affect the learning behaviors in SAM R1. DX-9386 reduced the elevated levels of lipid peroxide in the serum and liver of SAM P8, while it did not alter that in SAM R1. These results suggested that DX-9386 slowed the aging process of SAM P8 in terms of learning behaviors and lipid peroxidation.
Ingredients of a Chinese Prescription DX-9386, Individually Promote Hippocampal Long-Term Potentiation in Vivo
DX-9386 is a traditional Chinese medicinal prescription consisting of ginseng (Panax Ginseng C. A. MEYER), polygala (Polygala Tenuifolia WILLDENEW), acorus (Acorus Gramineus SOLAND) and hoelen (Poria Cocos WOLF). We recently found that oral administration of the prescription at a dose of 500 mg/kg intensified the formation of long-term potentiation (LTP) in the dentate gyrus of anesthetized rats. To evaluate the individual contribution of separate ingredients in DX-9386 towards the observed biological activity, we investigated their direct influence upon LTP formation in vivo. A single oral administration of hoelen and ginseng (250 and 500 mg/kg) significantly increased the spike amplitude evoked by a subthreshold tetanic stimulation at time intervals up to 30 min after tetanus. Only minor effects of polygala (500 mg/kg) and no influence of acorus up to 500 mg/kg were observed. No drugs affected the basal spike amplitude induced by a test stimulus. In addition, we ascertained that DX-9386 was also active at a dose of 250 mg/kg. Taken together, these results indicate that hoelen and ginseng are the active components of DX-9386 with regard to the enhancement of hippocampal LTP.
(Commonly known as “Sweet Flag”)
Phenolic constituents from the rhizomes of Acorus gramineus and their biological evaluation on antitumor and anti-inflammatory activities
On the search for anti-cancer compounds from natural Korean medicinal sources, a bioassay-guided fractionation and chemical investigation of the MeOH extract from the rhizomes of Acorus gramineus resulted in the isolation and identification of thirteen phenolic derivatives (1–13) including two new 8-O-4′-neolignans, named surinamensinols A (1) and B (2) and a new phenolic compound, named acoramol (9). The structures of these new compounds were elucidated on the basis of 1D and 2D NMR spectroscopic data analyses as well as circular dichroism (CD) spectroscopy studies. The cytotoxic activities of the isolates (1–13) were evaluated by determining their inhibitory effects on human tumor cell lines. The new 8-O-4′-neolignans, compounds 1 and 2, showed moderate antiproliferative activities against A549, SK-OV-3, SK-MEL-2, and HCT-15 cell lines with IC50 values in the range of 4.17–26.18 μM. On the basis of the expanded understanding that inflammation is a crucial cause of tumor progression, anti-inflammatory activities of these compounds were determined by measuring nitric oxide (NO) levels in the medium using murine microglia BV-2 cells. Compounds 1, 2, 4, 7 and 10 inhibited NO production in BV-2 stimulated by lipopolysaccharide with IC50 values of 8.17–18.73 μM via NO scavenging, inhibition of iNOS activity, and/or suppression of iNOS expression.
Immuno-regulatory and Anti-cancer Effect of Acorus gramineus Solander
Methanol extracts of Acorus gramineus Solander(AGS) were found to exhibit immuno-regulatory action in BALB/c mice. Oral administration of AGS increased murine splenic T lymphocytes, especially TH and TC/TS subpopulations were increased significantly. Treatment of AGS exerted strong cytotoxicity against U937 and HL60 human leukemia cells. Also, AGS induced apoptosis of U937 leukemia cells in a dose dependent manner. Nitric oxide(NO) production and iNOS gene expression were also increased in AGS-treated RAW264.7 cells. Treatment of AGS increased the expression of p53 gene and decreased the expression of PCNA protein in cultured U937 cells. These data suggest that AGS are effective on the immuno-regulatory action and anti-cancer properties.
Acetylcholinesterase-inhibitory and memory-enhancing effect
In Ayurveda, herbal medicines with rasayana. effects are believed to be restorative, to attain longevity, intelligence, and freedom from age-related disorders. AC is regarded in Ayurvedic medicine as promoting rasayana effects and has been used to treat memory loss (Kirtikar & Basu, 1954; Mukherjee & Wahile, 2006a). AC is used in Ayurvedic medicine on a regular basis for the treatment of memory loss and other mental disorders (Kirtikar & Basu, 1954; Howes & Houghton., 2003). AC extract has also been used as traditional Chinese prescription, and its beneficial effects on memory disorder, on learning performance, lipid peroxide content, and anti-aging effects in senescence have been reported (Nishiyama et al., 1994; Zhang et al., 1994). The in vitro. acetylcholinesterase (AChE) inhibitory effect of hydro- alcohol extract and essential oil of AC rhizomes was reported based on Ellman’s method in 96-well microplates using bovine erythrocytes. The essential oil showed stronger inhibition than the hydro-alcohol extract (Houghton et al., 2006a). Methanol extracts of AC showed significant acetylcholinesterase enzyme inhibition at a concentration 200 µg/mL (Oh et al., 2004). Houghton et al. (2006b) reported the in vitro. acetylcholinesterase inhibitory effect of β.-asarone and α.-asarone. β.-Asarone isat least an order of magnitude more active than its trans.-isomer α.-asarone. The AChE-inhibitory activity of the oil can be ascribed to β.-asarone. Because cognitive performance and memory are related to acetylcholine levels, the AChE-inhibitory effect of the plant may account for its traditional use.
Suppresses NF-kB
We revealed that β-Asarone (found in Acorus) suppressed not only basal NF-κB activity (activated in aging) but also Tumor necrosis factor α (TNF-α) induced NF-κB activity. Moreover, blocking NF-κB signaling inactivation was critical for β-Asarone induced apoptosis and inhibition of proliferation.

Polgala Tenuifolia
INHIBTS TLR4 & NFKB
Anti-inflammatory effects of Polygala tenuifoliaroot through inhibition of NF-κB activationin lipopolysaccharide-induced BV2 microglial cells
The root of Polygala tenuifolia Willd is a well-known traditional Oriental medicine and has been prescribed for treatment of dysfunction in memorial systems and various brain inflammatory diseases. The present study was designed to validate the anti-inflammatory effects of the water extract of Polygala tenuifolia root (WEPT).
Materials and methods: The anti-inflammatory properties of WEPT were studied using lipopolysaccharide (LPS)-stimulated murine BV2 microglia model. As inflammatory parameters, the production of nitric oxide (NO), inducible NO synthase (iNOS), cyclooxygenase (COX)-2, prostaglandin E2 (PGE2), tumor necrosis factor (TNF)-α, and interleukin (IL)-1β were evaluated. We also examined the extract’s effect on the activity of nuclear factor-kappaB (NF-κB), and toll-like receptor 4 (TLR4) and myeloid differentiation factor 88 (Myd-88) expression.
Results: WEPT suppressed LPS-induced production of NO, PGE2, and expression of iNOS and COX-2 in a dose-dependent manner, without causing cytotoxicity. It also significantly reduced generation of proinflammatory cytokines, including IL-1β and TNF-α. In addition, WEPT suppressed NF-κB translocation by blockade of IkappaB-α (IκB-α) degradation and inhibited TLR4 and Myd-88 expression in LPS-stimulated BV2 cells.
Conclusions: These results indicate that the inhibitory effects of WEPT on LPS-stimulated inflammatory mediator production in BV2 microglia are associated with suppression of the NF-κB and toll-like receptor signaling pathways. Therefore, Polygala tenuifolia extracts may be useful in treatment of neurodegenerative diseases by inhibition of inflammatory mediator production in activated microglia.

HOELEN (Poria Cocos)
downregulateS AKT & C-FLIP
Activated Akt signaling in cancer cells inhibits apoptosis by phosphorylation of downstream substrates, such as BAD and Bcl-2, which are involved in the regulation of the intrinsic apoptotic pathway (Vivanco and Sawyers 2002). Furthermore, Akt also inhibits the death receptor-mediated apoptosis pathway through up-regulation of c-FLIP expression (Panka et al. 2001). As an anti-apoptotic protein, c-FLIP blocks apoptosis induced by the oligomerization of the adapter protein and therefore functions as a caspase-8 dominant negative (Panka et al. 2001). Targeting the PI3-kinase/Akt signaling pathway has been reported to be an effective strategy for the treatment of lung cancer (Crowell and Steele 2003). In the experiments, PPAC treatment decreased activation of Akt and increased the stability of PTEN in A549 cells. Based on these results, it was therefore postulated that the down-regulation of Akt activitymight decrease c-FLIP expression, which would in turn cause the activation of caspase-8. The suppression of Akt activity by PPAC might be one of the essential mechanisms of PPAC-induced apoptosis. However, it remains to be elucidated whether Akt inhibition would solely or in combination with other factors contribute towards PPAC-induced apoptosis.

EF24 (Tumeric Root)
(curcumin analog. More potent and bioavailable than curcumin, with 10-fold greater potency).
novel Senolytic agent
NF-KB Inhibitor
IKK inhibitor
Bioactivities of EF24, a Novel Curcumin Analog: A Review
Inhibition of NF-κB Signaling
Most studies suggest that EF24 impairs cell growth by inducing cell cycle arrest followed by induction of apoptosis, which is accompanied by caspase-3 activation. However, the cell signaling pathway mediating the EF24 effect was not elucidated until 2008 when Kasinski et al. first revealed that EF24 induced cell apoptosis via suppressing NF-κB signaling pathway through direct action on IkB kinase (IKK) (21). NF-κB regulates a wide variety of genes involved in cell proliferation, differentiation, cell cycle control (36), oncogenic activation (37) and metastasis (38). EF24 can inhibit the catalytic activity of IKK protein complex, which blocks IκB phosphorylation and subsequent degradation, and finally prevents the nuclear translocation of p65 subunit of NF-κB. The study provides a molecular explanation for the superior activity of EF24 over curcumin (with a potency about 10 times higher than that of curcumin). Other groups further verified the involvement of EF24 in inhibiting NF-κB signaling pathway in cancer (39–42). EF24 robustly conferred radiation-induced cell death mainly by inhibiting radiation-induced NF-κB signaling in breast cancer (43). In the same year, the same group found that EF24 can suppress the radiation-induced NF-κB-DNA binding activity/promoter activation in genetically varied human neuroblastoma (44). Inhibition of the NF-κB signaling extends the therapeutic application of EF24 to other NF-κB-dependent diseases, such as inflammatory diseases (described below).
Comparison of EF24 to Curcumin in Anti-cancer Activity and Bioavailability
Since the development of EF24, scientists have compared its activity with its parent compound curcumin. Most of the studies were focused on the anti-cancer activity. Adams et al. synthesized a series of curcumin analogs including EF24, and submitted them to the NCI anti-cancer cell line screen. The results showed that EF24 was effective against all of the cell lines. The mean panel GI50 (concentration at which the drug inhibits tumor cell growth by 50%) was 10-fold better than curcumin and cisplatin (18). The authors also submitted the analogs to an in vitro anti-angiogenesis screen and revealed that EF24 was more active than curcumin in the assay. Their in vivo experiment suggested that EF24 was well tolerated by mice and much safer than the chemotherapy drug cisplatin (18). Later, Subramaniam et al. further compared the potency of EF24 to curcumin in gastrointestinal cancer cells and demonstrated that EF24 was more potent than curcumin. For example, 1 μmol/L of EF24 significantly suppressed proliferation and colony formation of the colon and gastric cancer cell lines while at the same dose of curcumin had no effect (29). Consistently, EF24 exhibited IC50 values 10 to 20 times lower than that of curcumin in multiple cancer cell lines, including lung, ovarian, cervical, breast, prostate cancer cells and cholangiocarcinoma cells (21, 22, 28, 40, 48). Similarly, in human osteogenic sarcoma cells (Saos2), EF24 was 3-fold more potent than curcumin (31). This evidence collectively suggests that EF24 displays much more potent anti-cancer activity than its parent compound in vitro. Although EF24 shared many anti-cancer mechanisms with its parent compound curcumin, such as inhibiting NF-κB and HIF-1α, it exerts its effects in different ways, for example, in how it regulates HIF-1α activity (described above). In addition, curcumin efficiently inhibited proteasome activity. By contrast, EF24 was 20-fold less active than curcumin for proteasome inhibition (45). Likewise, curcumin can regulate the STAT3, while EF24 has no effect on STAT activation (64).
The in vivo activity of a compound relies on the bioavailability of the compound at the site of the tumor. Dietary curcumin is poorly absorbed through the intestinal tract, therefore curcumin does not have a therapeutic effect at low doses (29). By contrast, EF-24 has higher oral bioavailability (60%) in mice (66), explaining to some extent the improved in vivo activity of EF24 compared to curcumin.
In addition to the improvement of anti-cancer activity, EF24 shows low toxicity to normal cells. EF24 has been shown to induce apoptosis in cancer cells and inhibit the growth of human breast tumors in a mouse xenograft model but showed low toxicity (18). Subramaniam et al. reported that EF24 inhibited intestinal cancer cell proliferation, but did not affect the proliferation of normal mouse embryonic fibroblasts cells, suggesting that EF24 is not toxic to normal cells (29). Similarly, EF24 inhibits tumor growth in human cholangiocarcinoma while displaying low toxicity levels. As a sensitizer, co-treatment of EF24 and rapamycin selectively enhances the cytotoxicity in gastric cancer cells but not in normal cells (60). Above all, multiple molecular targets, wide-spectrum potency, enhanced bioavailability as well as low toxicity to normal cells confer EF24 a series of advantages in clinical applications.
Regulation of HIF-1α Expression
Another important role of EF24 is to regulate HIF-1α expression which is closely associated with the outcome of chemotherapy in cancer treatment. For example, Liang et al. reported that sorafenib therapy could induce drug resistance in the treatment of hepatocellular carcinoma, in which HIF-1α plays an important role (50). Anti-angiogenic effects of sustained sorafenib therapy caused intratumor hypoxia, which induced HIF-1α and protected cancer cells from sorafenib treatment. EF24 can inhibit HIF-1α by sequestering it in cytoplasm and can promote its degradation by upregulating Von Hippel-Lindau tumor suppressor (VHL). Combination of EF24 and sorafenib showed synergistic effects against metastasis both in vivo and in vitro (50), providing compelling evidence for the potential of clinical application of EF24. Similar to EF24, the parent compound curcumin can also regulate HIF-1α expression (51), albeit through distinct mechanisms. Curcumin inhibited HIF-1α gene transcription, while EF24 exerted the activity by inhibiting HIF-1α post-transcriptionally (22).
The main mechanisms of action for EF24 include inhibition of the NF-κB pathway and HIF-1α protein and regulation of the MAPK pathway and ROS production.
downregulates cFLIP
cFLIP expression is regulated by a broad spectrum of mechanisms, including histone acetylation [32] DNA damage and ubiquitination [33] miR-375 [34] sonic hedgehog and myc signalling [32, 35]; and Hsp90 [36, 37]. Here we focussed on NF-κB-driven cFLIP expression because it is common in many cancers [21, 38] Indeed, the NF-κB inhibitor, EF24 elicited cFLIP downregulation in primary thymoma TECs and 1889 thymic carcinoma cells [13] (Figure and Supplementary Figure 6A) and sensitized them to TNFα induced cell death (Figure and and Supplementary Figure 6B and 6C). Like cFLIP knockdown, NF-κB blockade induced both apoptosis and autophagy in 1889c TC cells (Supplementary Figure 7 and Figure ) and in primary thymoma-derived TECs (Figure ; Figure and Figure ). Whether NF-κB expression in neoplastic TECs is driven by oncogenic mechanisms [38] or echoes the epithelial NF-κB signaling that is operative during early thymus development [39] is unknown.

EGCG
Suppression of NFkB and STAT3
Persistent activation of signal transducer and activator of transcription (STAT-3) is a common feature of PCa (20,21) and increased resistance to apoptosis occurs due to constitutive activation of STAT-3. Western blot analysis was performed to look at the protein expression of STAT-3, we observed a consistent upregulation of the protein as the tumor progressed from not-detectable at 8 weeks to poorly-differentiated at 32 weeks in dorsolateral prostate of TRAMP mice. This upregulation was significantly inhibited in age matched mice with continuous GTP infusion (). These results were further confirmed by immunohistochemical analysis of STAT-3 levels (), indicating a significant decrease in STAT-3 protein expression in green tea polyphenol-fed TRAMP mice.
Taken together this study highlights the chemo-preventive effects of oral infusion of GTP in TRAMP mice. We suggest that the observed inhibition of NFκB signaling pathway along withSTAT3 inhibition may potentially be involved in repressing tumor progression in TRAMP mice.
EGCG-induced JNK activation
Polyphenols such as epigallocatechin-3-gallate (EGCG) from green tea extract can exert a growth-suppressive effect on human pancreatic cancer cells in vitro. In pursuit of our investigations to dissect the molecular mechanism of EGCG action on pancreatic cancer, we observed that the antiproliferative action of EGCG on pancreatic carcinoma is mediated through programmed cell death or apoptosis as evident from nuclear condensation, caspase-3 activation and poly-ADP ribose polymerase (PARP) cleavage. EGCG-induced apoptosis of pancreatic cancer cells is accompanied by growth arrest at an earlier phase of the cell cycle. In addition, EGCG invokes Bax oligomerization and depolarization of mitochondrial membranes to facilitate cytochrome c release into cytosol. EGCG-induced downregulation of IAP family member X chromosome linked inhibitor of apoptosis protein (XIAP)might be helpful to facilitate cytochrome c mediated downstream caspase activation. On the other end, EGCG elicited the production of intracellular reactive oxygen species (ROS), as well as the c-Jun N-terminal kinase (JNK) activation in pancreatic carcinoma cells. Interestingly, inhibitor of JNK signaling pathway as well as antioxidant N-acetyl-L-cysteine (NAC) blocked EGCG-induced apoptosis. To summarize, our studies suggest that EGCG induces stress signals by damaging mitochondria and ROS-mediated JNK activation in MIA PaCa-2 pancreatic carcinoma cells.
The prevention of EGCG-mediated apoptosis by JNK inhibitor II and NAC suggests the involvement of ROS-mediated JNK activation in this pathway. The ROS comprise of singlet oxygen, hydroxyl radicals, superoxide, hydroperoxides and peroxides. In our studies, we have observed an increase in hydrogen peroxide level owing to EGCG exposure as noted in the case of lung tumor cell lines (21). Lei et al. (42) have indicated that JNK signaling is necessary for the stress-induced release of cytochrome c release and programmed cell death. JNK signaling and cytochrome c release may be interlinked to a pro-apoptotic member of Bcl-2 family because activated JNK is unable to evoke apoptosis in cells deficient of Bax. Our observation indicates that the concerted efforts of JNK activation and Bax oligomerization might play a pivotal role in the demise of pancreatic cancer cells.
Since increased ROS levels were observed to exert the activation of stress kinase JNK (40,41), we were interested to assess the activation status of JNK by immunocomplex kinase assay in the control and EGCG-exposed MIA PaCa-2 cells. Figure 10A clearly demonstrates the EGCG-induced JNK activation in MIA PaCa-2 cells. In order to understand whether JNK activation is necessary for EGCG-mediated cell killing, we next examined whether the blocking of JNK activity exerts any effect on EGCG-induced apoptosis.
BCL, Bcl-xL, Bcl-w INHIBITOR
high concentration of EGCG exerted a decrease in the expression of Bcl-xL and Bcl-w
MDM2 Inhibitor
Epigallocatechin gallate promotes p53 accumulation and activity via the inhibition of MDM2-mediated p53 ubiquitination in human lung cancer cells.
Epigallocatechin gallate (EGCG), which is derived from green tea, is well known for its chemopreventive activity. Several studies have shown that p53 plays an important role in the activity of EGCG; however, the mechanism by which EGCG regulates p53 requires further investigation. In the present study, we showed that EGCG inhibits anchorage-independent growth of human lung cancer cells by upregulating p53 expression. EGCG treatment can substantially increase p53 stability, promote nuclear localization of p53 and decrease nuclear accumulation of MDM2. We also found that EGCG increases the phosphorylation of p53 at Ser15 and Ser20 and enhances its transcriptional activity. Although EGCG promotes MDM2 expression in a p53-dependent manner, the interaction between MDM2 and p53 was significantly inhibited following EGCG treatment, which resulted in the inhibition of MDM2-mediated p53 ubiquitination. Thus, our results suggest that the stabilization and activation of p53 may partly contribute to the anticancer activity of EGCG.
SENOLYTIC ATTRIBUTES OF [EGCG] MATCHA
– Senescence is defined the condition or process of deterioration with age [177]. Senescence plays a role in multiple biological events, such as embryogenesis, tissue regeneration and repair, ageing, cardiovascular, renal, liver diseases, and cancer [178,179]. EGCG has been implicated in senescence in different works. For example human mesenchymal stem cells (hMSCs) show the peculiar behavior of producing abundant ROS during culturing and this issue has been exploited with a natural anti‐ oxidant as EGCG [180]. The work explored the anti‐senescent effect of EGCG in H2O2‐exposed hMSCs and proved that EGCG 50 or 100 μM reversed oxidative stress by downregulating the p53–p21 signaling pathway and upregulating Nrf2 expression. Similarly, in primary cells, including rat vascular smooth muscle cells (RVSMCs), human dermal fibroblasts (HDFs) and human articular chondrocytes (HACs), the same concentrations of EGCG (50 or 100 μM) could prevent senescence and recover cell cycle progression, again impacting on the p53 pathway at least in HDFs cells [181]. Furthermore, in 3T3‐L1 preadipocytes, EGCG could inhibit stress‐induced senescence (H2O2‐ induced) by countering DNA damage and cell cycle arrest [182]. In this work, EGCG (same concentrations as above, 50 or 100 μM) modulated PI3K/Akt/mTOR and AMPK pathways by blocking ROS, iNOS, Cox‐2, NF‐kB, and p53, and increasing apoptosis by suppressing antiapoptotic protein Bcl‐2. Moreover, in WI‐38 fibroblasts at late‐stage (population doubling > 25) used as model of oxidative stress and inflammation, EGCG (25, 50, 100 μM) reduced TNF‐α, IL‐6, ROS, again acting on p53 [183]. ECGC also decreased retinoblastoma expressions, while enhancing E2F2 expressions and superoxide dismutase (SOD) 1 and 2, all being implicated in oxidation processes. EGCG has been shown to affect the development and severity of cellular senescence, but from a therapeutic perspective more work is needed to justify its implication in human consumption. 💥Senolytic attributes of EGCG are interesting, especially for attenuating age‐associated disorders, though clinical trails and health consequence still need to be proposed and discovered.
TLR4 INHIBITOR
TLR4 signaling inhibitory pathway induced by green tea polyphenol epigallocatechin-3-gallatethrough 67-kDa laminin receptor.
Epigallocatechin-3-gallate (EGCG), a major active polyphenol of green tea, has been shown to downregulate inflammatory responses in macrophages; however, the underlying mechanism has not been understood. Recently, we identified the 67-kDa laminin receptor (67LR) as a cell-surface EGCG receptor that mediates the anticancer action of EGCG at physiologically relevant concentrations (0.1-1 microM). In this study, we show the molecular basis for the downregulation of TLR4 signal transduction by EGCG at 1 microM in macrophages. Anti-67LR Ab treatment or RNA interference-mediated silencing of 67LR resulted in abrogation of the inhibitory action of EGCG on LPS-induced activation of downstream signaling pathways and target gene expressions. Additionally, we found that EGCG reduced the TLR4 expression through 67LR. Interestingly, EGCG induced a rapid upregulation of Toll-interacting protein (Tollip), a negative regulator of TLR signaling, and this EGCG action was prevented by 67LR silencing or anti-67LR Ab treatment. RNA interference-mediated silencing of Tollip impaired the TLR4 signaling inhibitory activity of EGCG. Taken together, these findings demonstrate that 67LR plays a critical role in mediating anti-inflammatory action of a physiologically relevant EGCG, and Tollip expression could be modulated through 67LR. These results provide a new insight into the understanding of negative regulatory mechanisms for the TLR4 signaling pathway and consequent inflammatory responses that are implicated in the development and progression of many chronic diseases.
EMBELIN
STAT3 INHIBITOR
Even though embelin, an inhibitor of the XIAP, is known to exhibit anti-inflammatory and anti-cancer activities, very little is known about its mechanism of action. Here, we investigated whether embelin mediates its effect through interference with the signal transducer and activator of transcription 3 (STAT3) pathway. We found that embelin inhibited constitutive STAT3 activation in a variety of human cancer cell lines such as U266, DU-145, and SCC4 cells. The suppression of STAT3 was mediated through inhibition of the activation of JAK2 and c-Src. Pervanadate treatment also reversed the embelin-induced down-regulation of STAT3, suggesting the involvement of a protein tyrosine phosphatase. Indeed, we found that embelin-induced the expression of the tyrosine phosphatase PTEN and deletion of the PTEN gene by small interfering RNA abolished the ability of embelin to inhibit STAT3 activation. Besides, embelin failed to suppress STAT3 activation in PTEN-null PC3 cells, thus indicating that the inhibitory effect of embelin on STAT3 is PTEN-dependent. Embelin down-regulated the expression of STAT3-regulated gene products; this correlated with the suppression of cell proliferation and invasion, and the induction of apoptosis through the activation of caspase-3. Overall, our results indicate that the anti-inflammatory and anti-cancer activities previously assigned to embelin may be mediated in part through the suppression of the STAT3 pathway.
► STAT3 plays an important role in carcinogenesis and metastasis of various human cancers. ► Embelin could suppress constitutive STAT3 activation through induction of PTEN. ► Embelin down-regulated the expression of STAT3-regulated gene products.
Embelin: a benzoquinone possesses therapeutic potential for the treatment of human cancer
Natural products have been gaining recognition and are becoming a significant part of research in the area of drug development and discovery. Phytochemicals derived from these sources have been comprehensively studied and have displayed a wide range of activities against many fatal diseases including cancer. One such product that has gained recognition from its pharmacological properties and nontoxic nature is embelin, obtained from Embelia ribes. Amid all the vivid pharmacological activities, embelin has gained its prominence in the area of cancer research. Embelin binds to the BIR3 domain of XIAP, preventing the association of XIAP and caspase-9 resulting in the suppression of cell growth, proliferation and migration of various types of cancer cells. Furthermore, embelin modulates anti-apoptotic pathways by suppressing the activity of NF-κB, PI3-kinase/AKT, JAK/STAT pathway– among others. The present review summarizes the various reported effects of embelin on different types of cancer cells and highlights the cellular mechanisms of action.
Emodin
Emodin has a cytotoxic activity against human multiple myeloma as a JAK 2 inhibitor.
Emodin is an active component of a traditional Chinese and Japanese medicine isolated from the root and rhizomes of Rheum palmatum L. Here, we show that emodin significantly induces cytotoxicity in the human myeloma cells through the elimination of myeloid cell leukemia 1 (Mcl-1). Emodin inhibited interleukin-6-induced activation of Janus-activated kinase 2 (JAK2) and phosphorylation of signal transducer and activator of transcription 3 (STAT3), followed by the decreased expression of Mcl-1. Activation of caspase-3 and caspase-9 was triggered by emodin, but the expression of other antiapoptotic Bcl-2 family members, except Mcl-1, did not change in the presence of emodin. To clarify the importance of Mcl-1 in emodin-induced apoptosis, the Mcl-1 expression vector was introduced into the human myeloma cells by electroporation. Induction of apoptosis by emodin was almost abrogated in Mcl-1-overexpressing myeloma cells as the same level as in parental cells, which were not treated with emodin. In conclusion, emodin inhibits interleukin-6-induced JAK2/STAT3 pathway selectively and induces apoptosis in myeloma cells via down-regulation of Mcl-1, which is a good target for treating myeloma. Taken together, our results show emodin as a new potent anticancer agent for the treatment of multiple myeloma patients.
Emodin enhances cytotoxicity of chemotherapeutic drugs in prostate cancer cells: the mechanisms involve ROS-mediated suppression of multidrug resistance and hypoxia inducible factor-1.
The intrinsic or acquired resistance to multiple drugs (MDR) of cancer cells remains one of the main obstacles for chemotherapy. Development of small molecule targeting to hypoxia inducible factor-1 (HIF-1) has been recently proposed as strategy for treatments of drug-resistant solid tumors. In the present study, emodin, proven as a reactive oxygen species (ROS) generator by our previous work, was applied in combination with cisplatin and other chemotherapeutic drugs in the multidrug resistant prostate carcinoma cell line DU-145 and normal human dermal fibroblasts. Results showed that emodin/cisplatin co-treatment remarkably elevated ROS level and enhanced chemosensitivity in DU-145 cells, compared with cisplatin-only treatment, but exerted little effect on non-tumor cells. The effect of co-treatment on MDR1 gene and its upstream regulator HIF-1 was then investigated in DU-145. Co-treatment downregulated MDR1 expression and promoted drug retention, and meanwhile suppressed transactivation of HIF-1 in response to hypoxia without changing expression of HIF-1 alpha. The experiments on tumor-bearing mice showed that co-treatment inhibited the tumor growth in vivo, owing to oxidative stress and MDR1 down-regulation within tumors. HIF-1 transactivation and clonegenesis were suppressed in cells isolated from the tumors. Finally, examinations for the body weight, the organ histology and the antioxidant capacity of serum suggested that no systemic toxicity related to co-treatment was discernable. In conclusions, emodin, as a novel small inhibitor of HIF-1, may be recognized an effective adjunctive to improve efficacy of cytotoxic drugs in prostate cancer cells with over-activated HIF-1 and potent MDR.
Matrix metalloproteinases (MMPs) are the proteases involved in the degradation of the extracellular matrix. MMP-1 is thought to be one of the key enzymes acting in fibrolysis, a process closely related to tissue remodeling. In this study, we found that emodin, an anthraquinone which has been isolated from the rhizome of Rheum palmatum, significantly inhibited TNF alpha-induced MMP-1 gene expressionin a concentration-dependent manner. Therefore, we have attempted to characterize the inhibitory mechanism of emodin in TNF alpha-induced MMP-1 expression. Emodin was determined to inhibit TNF alpha-induced activation of AP-1 promoter, an important nuclear transcription factor in MMP-1 expression. Additionally, we detected that emodin suppressed the TNF alpha-induced phosphorylation of two mitogen-activated protein kinases, extracellular signal-regulated protein kinase and c-Jun N-terminal kinase, but it did not suppress the TNF alpha-induced phosphorylation of p38 kinase. In a consistent result, the TNF alpha-induced MMP-1 expression was inhibited by PD98059 (MEK/ERK inhibitor) and SP600125 (JNK inhibitor), but was not inhibited by SB203580, a p38 MAPK inhibitor. Taken together, these results show that emodin suppresses TNF alpha-induced MMP-1 expression through the inhibition of the AP-1 signaling pathway.
CASPASE 3 ACTIVATOR
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is an active constituent of Rheum palmatum, and showed inhibitory activity on lipopolysaccharide-induced NO production in our previous study. However, the apoptosis-inducing activity of emodin has remained undefined. Among three structurally related anthraquinones, including emodin, physcion, and chrysophanol, emodin showed the most potent cytotoxic effects on HL-60 cells, accompanied by the dose- and time-dependent appearance of characteristics of apoptosis including an increase in DNA ladder intensity, morphological changes, appearance of apoptotic bodies, and an increase in hypodiploid cells. Emodin at apoptosis-inducing concentrations causes rapid and transient induction of caspase 3/CPP32 activity, but not caspase 1 activity, according to cleavage of caspase 3 substrates poly(ADP-ribose) polymerase and D4-GDI proteins, the appearance of cleaved caspase 3 fragments being detected in emodin- but not physcion- or chrysophanol-treated HL-60 cells. A decrease in the anti-apoptotic protein, Mcl-1, was detected in emodin-treated HL-60 cells, whereas other Bcl-2 family proteins including Bax, Bcl-2, Bcl-XL, and Bad remained unchanged. The caspase 3 inhibitor, Ac-DEVD-CHO, but not the caspase 1 inhibitor, Ac-YVAD-CHO, attenuated emodin-induced DNA ladders, associated with the blockage of PARP and D4-GDI cleavage. Free radical scavenging agents including NAC, catalase, SOD, ALL, DPI, l-NAME and PDTC showed no preventive effect on emodin-induced apoptotic responses, whereas NAC, CAT and PDTC prevented HL-60 cells from ROS (H2O2)-induced apoptosis through inhibition of caspase 3 cascades. Induction of catalase, but not SOD, activity was detected in emodin-treated HL-60 cells by in gel activity assays, and H2O2-induced intracellular peroxide level was significantly reduced by prior treatment of emodin in HL-60 cells. Our experiments provide evidence that emodin is an effective apoptosis inducer in HL-60 cells through activation of the caspase 3 cascade, but that it is independent of ROS production.
Esculetin
CASPASE 3 ACTIVATOR
In this study, we have investigated whether esculetin exerts anti-proliferative and apoptotic effects on human leukemia U937 cells. It was found that esculetin could inhibit cell viability in a time-dependent manner, which was associated with the induction of apoptotic cell death such as increased populations of apoptotic- sub G1 phase. Apoptosis of U937 cells by esculetin was associated with an inhibition of Bcl-2/Bax binding activity, formation of tBid, down-regulation of X-linked inhibitor of apoptotic protein (XIAP) expression, and up-regulation of death receptor 4 (DR4) and FasL expression. Esculetin treatment also induced the degradation of {beta}-catenin and DNA fragmentation factor 45/inhibitor of caspase-activated DNase (DFF45/ICAD). Furthermore, a caspase-3 specific inhibitor, z-DEVD-fmk, significantly inhibited sub-G1 phase DNA content, morphological changes and degradation of {beta}-catenin and DEE45/ICAD. These results indicated that a key regulator in esculetin-induced apoptosis was caspase-3 in human leukemia U937 cells.
evodiamine
Atgs↑, 3-MA⊥, IL6↓, STAT3⊥, AP-1⊥, PLC- γ1⊥, XIAP⊥, Bax↑, CDK1⊥, ND cyclinB1↑, PI3K⊥, Akt⊥, PKA⊥, mTOR⊥, PTEN⊥, NF-κB↓, cyclinA↓, cyclinA-dependent kinase 2↓, cdc25c↓, TUNEL↑, procaspase-3-8-9↓, cdc25C↑, cyclin B1↑, cdc2-p161 protein↑, cdc2-p15, caspase-3-8-9↑, Fas-L↑, p53↑, p21↑, Bcl-2↓, TopI⊥, Raf-1↓, Bax↑, Bcl-2↑, Bcl-x(L)↓, Beclin 1↑, LC3↑, Cdc2↑, cyclin B1↑, Cdc2 (Thr 161) ↑, Cdc2 (Tyr 15) ↓, Myt-1↓, Cdc25C↓, caspase-3-9↑, ERK phosphorylation↓, VEGF⊥
Evodiamine (EVO) is an indoloquinazoline alkaloid that exerts its various anti-oncogenic actions by blocking phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt), mitogen-activated protein kinase (MAPK), c-Met, and nuclear factor kappa B (NF-κB) signaling pathways, thus leading to apoptosis of tumor cells. We investigated the ability of EVO to affect hepatocyte growth factor (HGF)-induced c-Met/Src/STAT3 activation cascades in castration-resistant prostate cancer (CRPC). First, we noted that EVO showed cytotoxicity and anti-proliferation activities in PC-3 and DU145 cells. Next, we found that EVO markedly inhibited HGF-induced c-Met/Src/STAT3 phosphorylation and impaired the nuclear translocation of STAT3 protein. Then, we noted that EVO arrested the cell cycle, caused apoptosis, and downregulated the expression of various carcinogenic markers such as B-cell lymphoma 2 (Bcl-2), B-cell lymphoma-extra large (Bcl-xL), cyclin D1, cyclooxygenase 2 (COX-2), survivin, vascular endothelial growth factor (VEGF), and matrix metallopeptidases 9 (MMP-9). Moreover, it was observed that in cPC-3 and DU145 cells transfected with c-Met small interfering RNA (siRNA), Src/STAT3 activation was also mitigated and led to a decrease in EVO-induced apoptotic cell death. According to our results, EVO can abrogate the activation of the c-Met/Src/STAT3 signaling axis and thus plays a role as a robust suppressor of tumor cell survival, proliferation, and angiogenesis.
Fisetin
(from Cotinus coggygria Scop. aka “smoke tree”)
Fisetin is a senotherapeutic that extends health and lifespan.
BACKGROUND: Senescence is a tumor suppressor mechanism activated in stressed cells to prevent replication of damaged DNA. Senescent cells have been demonstrated to play a causal role in driving aging and age-related diseases using genetic and pharmacologic approaches. We previously demonstrated that the combination of dasatinib and the flavonoid quercetin is a potent senolytic improving numerous age-related conditions including frailty, osteoporosis and cardiovascular disease. The goal of this study was to identify flavonoids with more potent senolytic activity.
METHODS: A panel of flavonoid polyphenols was screened for senolytic activity using senescent murine and human fibroblasts, driven by oxidative and genotoxic stress, respectively. The top senotherapeutic flavonoid was tested in mice modeling a progeroid syndrome carrying a p16INK4a-luciferase reporter and aged wild-type mice to determine the effects of fisetin on senescence markers, age-related histopathology, disease markers, health span and lifespan. Human adipose tissue explants were used to determine if results translated.
FINDINGS: Of the 10 flavonoids tested, fisetin was the most potent senolytic. Acute or intermittent treatment of progeroid and old mice with fisetin reduced senescence markers in multiple tissues, consistent with a hit-and-run senolytic mechanism. Fisetin reduced senescence in a subset of cells in murine and human adipose tissue, demonstrating cell-type specificity. Administration of fisetin to wild-type mice late in life restored tissue homeostasis, reduced age-related pathology, and extended median and maximum lifespan.
INTERPRETATION: The natural product fisetin has senotherapeutic activity in mice and in human tissues. Late life intervention was sufficient to yield a potent health benefit. These characteristics suggest the feasibility to translation to human clinical studies.
New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463.
Senescent cells accumulate with aging and at sites of pathology in multiple chronic diseases. Senolytics are drugs that selectively promote apoptosis of senescent cells by temporarily disabling the pro-survival pathways that enable senescent cells to resist the pro-apoptotic, pro-inflammatory factors that they themselves secrete. Reducing senescent cell burden by genetic approaches or by administering senolytics delays or alleviates multiple age- and disease-related adverse phenotypes in preclinical models. Reported senolytics include dasatinib, quercetin, navitoclax (ABT263), and piperlongumine. Here we report that fisetin, a naturally-occurring flavone with low toxicity, and A1331852 and A1155463, selective BCL-XL inhibitors that may have less hematological toxicity than the less specific BCL-2 family inhibitor navitoclax, are senolytic. Fisetin selectively induces apoptosis in senescent but not proliferating human umbilical vein endothelial cells (HUVECs). It is not senolytic in senescent IMR90 cells, a human lung fibroblast strain, or primary human preadipocytes.A1331852 and A1155463 are senolytic in HUVECs and IMR90 cells, but not preadipocytes. These agents may be better candidates for eventual translation into clinical interventions than some existing senolytics, such as navitoclax, which is associated with hematological toxicity.
Fisetin activates Hippo pathway and JNK/ERK/AP-1 signaling to inhibit proliferation and induce apoptosis of human osteosarcoma cells via ZAK overexpression.
Osteosarcoma (OS) is a tumor entity that can cause a large number of cancer-related deaths. Although chemotherapy can decrease proliferation and increase apoptosis of human OS cells, the clinical prognosis remains poor. Fisetin is a flavonol found in fruits and vegetables and is reported to inhibit cell growth in numerous cancers. But the molecular mechanism underlying fisetin in human OS cells is not clear. It is known that sterile-alpha motif and leucine zipper containing kinase (ZAK), a kinase in the MAP3K family, is involved in various cell processes, including proliferation and apoptosis. In our lab, we have demonstrated that overexpression of ZAK can induce apoptosis in human OS cells. In the previous studies, MAP4K, the upstream of MAP3K, can act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Turning on the Hippo pathway can decrease proliferation and otherwise cause cell apoptosis in cancer cells. In this study, we found that fisetin can upregulate ZAK expression to induce the Hippo pathway and mediate the activation of JNK/ERK, the downstream of ZAK, to trigger cell apoptosis via AP-1 dependent manner in human OS cells. These findings reveal a novel molecular mechanism underlying fisetin effect on human OS cells.
MTOR INHIBITOR
Fisetin (3,7,3′,4′-tetrahydroxyflavone) belongs to the flavonol subgroup of flavonoids which includes quercetin, myricetin and kaempferol. Epidemiological and preclinical studies have shown that fisetin consumption affects various molecular targets which collectively slow the ageing process (George, 2016). Fisetin has been shown to inhibit the association between mTOR pathway signaling pathway constituents, Raptor, and Rictor. Fisetin also decreases PI3-K content, and enhances phosphorylation of Akt. Phosphorylated Akt in turn inhibits mTOR activity. Fisetin can also directly activate negative regulators of mTOR such as Tsc complex and AMPK as shown in Fig. 1 (Adhami et al., 2012). Moreover, fisetin has been shown to enhance cytotoxic effects when used in combination with other chemotherapeutic drugs (Haddad et al., 2010; Klimaszewska-Wisniewska et al., 2016).
inhibition of GSK3β
In summary, our definitive and novel findings obtained in this study indicate that fisetin confers cardioprotection against myocardial IRI, by bolstering the mitochondrial physiology, suppressing the oxidative stress, and augmenting the mitochondrial biogenesis, and these effects are mediated via inhibition of GSK3β activity. Since fisetin is well tolerated in human subjects and does not show toxic effects, this natural small molecule has bright prospects for further pharmaceutical development to be used against I/R-induced myocardial tissue injury and potentially for the treatment of cardiovascular diseases.
BCL-W INHIBITOR
This study aimed to investigate the effects of fisetin, a common dietary natural flavonoid, on apoptosis of Huh-7 cells. The MTT assay was used to evaluate the reduction of cell viability. In the DNA fragmentation assay and comet assay, fisetin-induced DNA damage was visible as a formation of DNA fragmentation and comet tails. Fisetin also induced intracellular accumulation of reactive oxygen species. Two-dimensional gel electrophoresis was performed to evaluate protein expression in fisetin-treated and control cells, and Baculoviral IAP repeat-containing protein 8 (BIRC8) and apoptosis regulator Bcl-W (Bcl2L2) were identified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Their expression was confirmed by western blotting. The anticancer effect of fisetin in Huh-7 cells may result from the down regulation of BIRC8 and the Bcl2L2.
enhancement of proapoptotic Bad and Bim
Although fisetin attenuates the COX-2 and MAPK pathways in HT-29 cells, the growth of HCT-116 cells was inhibited through the apoptosis mechanism. Several factors were involved in this mechanism, such as reduction of antiapoptotic Bcl-xL and Bcl-2 at the protein level and enhancement of proapoptotic Bad and Bim. Moreover, excitation of mitochondrial permeability and consequently activation of the caspase cascade, including caspase-3, -7, -8, and -9, and downstream factors such as cytochrome c are other means of promoting apoptosis. Activation of death receptors (Fas and tumor necrosis factor) by fisetin assisted in promoting apoptosis. In addition, fisetin increased p53 expression 32.
FOXO4 D-Retro-Inverso peptide
Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging
The accumulation of irreparable cellular damage restricts healthspan after acute stress or natural aging. Senescent cells are thought to impair tissue function and their genetic clearance can delay features of aging. Identifying how senescent cells avoid apoptosis allows for the prospective design of anti-senescence compounds to address whether homeostasis can also be restored.
Here, we identifyFOXO4 as a pivot in senescent cell viability. We designed a FOXO4 peptide which perturbs the FOXO4 interaction with p53. In senescent cells, this selectively causes p53 nuclear exclusion and cell-intrinsic apoptosis. Under conditions where it was well tolerated in vivo, this FOXO4 peptide neutralized Doxorubicin-induced chemotoxicity. Moreover, it restored fitness, fur density and renal function in both fast aging XpdTTD/TTD and naturally aged mice. Thus, therapeutic targeting of senescent cells is feasible under conditions where loss of health has already occurred and in doing so tissue homeostasis can effectively be restored.
FOXO4-DRI has a strong preference for targeting high-SASP subpopulations ofsenescent cells, but it is unclear what causes heterogeneity in the SASP. It will be a major achievement to unravel those mechanisms and to steer these such that therapeutic targeting is most beneficial. In that sense, identification of senescence-driven pathologies that rely on SASP may help in optimizing candidates for therapy. XpdTTD/TTD is pleiotropic model for aging that can be effectively used as a basis for such research. It is a well-established model for osteoarthritis, especially in cohorts of older age than we used here (52w) (Botter et al., 2011) and for the unhealthy loss in muscle (sarcopenia) and fat mass(Wijnhoven et al., 2005).
Last, it is relevant to note that independent of aging and age-related diseases, FOXO4-DRI may be of use against the progression, stemness and migration of malignant cancer. Given that SASP factors influence these(Campisi, 2013), it will be particularly interesting to determine whether FOXO4-DRI affects those p53-wt cancer cells that have adopted a more migratory and stem-like state due to reprogramming by chronic SASP exposure. In any case, the here reported beneficial effects of FOXO4-DRI provide a wide range of possibilities for studying the potential of therapeutic removal of senescence against diseases for which few options are available.
Regulation of cellular senescencevia the FOXO4‐p53 axis
Forkhead box O (FOXO) and p53 proteins are transcription factors that regulate diverse signalling pathways to control cell cycle, apoptosis and metabolism. In the last decade both FOXO and p53 have been identified as key players in aging, and their misregulation is linked to numerous diseases including cancers. However, many of the underlying molecular mechanisms remain mysterious, including regulation of ageing by FOXOs and p53. Several activities appear to be shared between FOXOs and p53, including their central role in the regulation of cellular senescence. In this review, we will focus on the recent advances on the link between FOXOs and p53, with a particular focus on the FOXO4‐p53 axis and the role of FOXO4/p53 in cellular senescence. Moreover, we discuss potential strategies for targeting the FOXO4‐p53 interaction to modulate cellular senescence as a drug target in treatment of aging‐related diseases and morbidity.
Given is role in aging and age‐related diseases, cellular senescence and associated signaling pathway have been extensively studied the past years. A better understanding of the exact molecular mechanisms involved in senescence is a key step toward the discovery of new drug that can efficiently and selectively target senescent cells in order to counteract age‐related pathology or more “utopically” in normal aging. In this way, we and co‐workers designed a peptide that interfere with FOXO‐p53 mediated senescence that showed great potency and selectivity in targeting apoptosis of senescent cells in mice. Based on this work, one can speculate on many other targets that could be used for similar therapeutic approaches. Indeed, interfering with (a) FOXO4 activation via the ROS/JNK or ROS/MDM2 pathway (b) nuclear translocation of active p53 (c) FOXO4 and/or p53 DNA binding to the promoter region of p21, could possibly lead to similar outcome.For this purpose, detailed structural information on binary complexes involved in the corresponding pathway are of great help in order to efficiently and selectively target one given interaction therefore limiting drug toxicity in vivo. Given the intricate networks in which p53 mediates the interaction with numerous binding partners, it remains to be studied how drug‐like molecules modulate binding of p53 to other p53 regulators. Therefore, atomic details of p53‐drug interactions are required in order to clarify the exact mechanisms leading to the clearance of senescent cells. Such structural and functional work will allow to optimize the binding properties, affinity/specificity of drug‐like molecules and to reduce potential off‐target effects.
In 2016, Van Deursen and coworkers showed that prophylactic ablation of cells that express the p16
Ink4a senescent marker mitigates tissue degeneration and extends the healthspan of mice (
Baker et al., 2016). Although the Baker et al. study established the causative role of senescent cells in tissue degeneration, it did not reveal the underlying mechanisms responsible for their generation. This is now addressed in the article by
Baar et al. (2017). The authors compared the expression profiles of normal human IMR90 cells with their radiation-induced senescent counterparts. Whereas they expected that the senescent cells would show reduced levels of pro-apoptotic and/or an increase in anti-apoptotic proteins, they observed the opposite, suggesting lack of an upstream trigger to activate the death program. Therefore, they searched for changes in the expression levels of transcription regulators that could underlie these changes. One immediate candidate was
FOXO4, a member of a transcription factor family previously implicated in aging and
longevity (
Martins et al., 2016), which appeared upregulated upon senescence induction. Therefore, FOXO4 was further scrutinized for its potential role in arresting cells in a senescent state. Indeed, inhibition of FOXO4 expression using a lentiviral
shRNA made cells undergo
apoptosis rather than become senescent upon irradiation. More importantly, FOXO4 inhibition in already senescent cells reduced their viability. This established that FOXO4 plays a critical role in consolidating the senescent state and that its inactivation causes senescent cells to undergo apoptosis.

FUCOIDAN
Caspase 3 Activator
Fucoidan, a sulfated polysaccharide in brown seaweed, was found to inhibit proliferation and induce apoptosis in human lymphoma HS-Sultan cell lines. Fucoidan-induced apop- tosis was accompanied by the activation of caspase-3and was partially prevented by pretreatment with a pan-caspase inhibitor, Z-VAD-FMK. The mitochondrial potential in HS-Sultan cells was decreased 24 hr after treatment with fucoidan, indicating that fucoi- dan induced apoptosis through a mitochondrial pathway. When HS-Sultan was treated with 100 mg/mL fucoidan for 24 hr, phosphorylation of ERK and GSK markedly decreased. In contrast, phosphorylation of p38 and Akt was not altered by treatment with fucoidan. L-Selectin and P-selectin are known to be receptors of fucoidan; however, as HS-Sultan does not express either of these selectins, it is unlikely that fucoidan induced apoptosis through them in HS-Sultan. The neutralizing antibody, Dreg56, against human L-selectin did not prevent the inhibitory effect of fucoidan on the proliferation of IM9 and MOLT4 cells, both of which express L-selectin; thus it is possible fucoidan induced apoptosis though different receptors. These results demonstrate that fucoidan has direct anti- cancer effects on human HS-Sultan cells through caspase and ERK pathways.

Studies have showed that most flavonoids exhibit anti-proliferative effects against tumor derived cell lines including leukemia[13], melanoma[14], colon[15], breast carcinoma[16], lung, and prostate[17]. Some reports have demonstrated that galangin is a naturally occurring non-toxic flavonoid with chemopreventive and anti-proliferative effects[6–8,18,19].
In this study, we demonstrated that galangin inhibited the proliferation of HCC cells and induced apoptosis at concentrations as low as 46.25 μmol/L and in excess of 185 μmol/L, respectively. Galangin-induced apoptosis was characterized by analyzing the effects on caspase-3 activation, PARP cleavage, and DNA condensation in HCC cells.
The mitochondrial pathway is commonly involved in the death stimuli. There are primarily two major events involved in apoptosis via the mitochondrial pathway. The first event is a change in mitochondrial membrane permeability, which leads to decreased mitochondrial membrane potential. Our data demonstrated reduced mitochondrial membrane potential as indicated by rhodamine 123 staining following treatment with galangin at different concentrations. The second event in the mitochondria-induced apoptotic pathway is the release of cytochrome c and AIF from the intermembrane space of the mitochondria into the cytosol. Here, we also showed that galangin increased the release of cytochrome c and AIF in the cytosol. Thus, our data indicates that galangin-induced apoptosis of HCC cells occurs through the mitochondrial pathway.
The Bcl-2 family of proteins is involved in the mitochondrial apoptotic pathway by inducing the release of cytochrome c from the mitochondrial intermembrane space. Cytochrome c cooperates with Apaf-1 (Apoptotic protease activating factor 1) to induce caspase activation, leading to cell apoptosis[20]. The Bcl-2 family proteins are categorized into three groups based on the four Bcl-2 homology domains (BH1-4 domains). Bcl-2, Bcl-w, Bcl-xL and Mcl-1, which contain BH domains 1-4 and are localized to the outer mitochondrial membrane, are anti-apoptotic Bcl-2 proteins[21]. These proteins can directly bind and inhibit the proapoptotic Bcl-2 family in the mitochondria pathway of apoptosis. The proapoptotic proteins of Bcl-2 family members are functionally divided into two classes. One class is the effector molecule, which includes Bak and Bax, and permeabilizes the outer mitochondrial membrane to release cytochrome c into the cytosol. The other class is the BH3-only proteins including Bad, Bid, Bik, Bim, Bmf, bNip3, Hrk, Noxa and Puma, which promote cell apoptosis through protein-protein interactions with other Bcl-2 family members[21].
Our data indicates that galangin causes Bax translocation to the mitochondria in HCC cells. In non-apoptotic cells, Bax is located in the cytosol or loosely bound to the outer membrane of the mitochondria in monomeric forms. However, Bax translocates to the mitochondrial membrane and homodimerizes in the presence of a death signal. As a result, the outer mitochondrial membrane is permeable to release cytochrome c and AIF into the cytosol. The release of cytochrome c, which is an important protein in the electron transfer chain, can lead to reduced mitochondria membrane potential and adenosine triphosphate (ATP) synthesis. The results of our experiments showed that galangin induces cytochrome c release and decreases mitochondrial membrane potential.
In the cytosol, cytochrome c can bind to Apaf-1, which is a cytosolic protein. Apaf-1 undergoes a conformational change upon binding to dATP or ATP, leading to the formation of the apoptosome complex. The apoptosome recruits procaspase-9, resulting in caspase 9-caspase 3 activation. This caspase cascade is responsible for the hydrolysis of key cytoplasmic proteins and for the cleavage of genomic DNA nucleosomes into 180 bp fragments via caspase-activated DNase, such as PARP. Caspase-3 is an executioner of apoptosis that subsequently cleaves many important intracelluar substrates, leading to chromatin condensation, nucleosomal DNA fragmentation, nuclear membrane breakdown, externalization of phosphatidylserine, and formation of apoptotic bodies[22]. Our data also shows galangin-treated HCC cells did indeed cause caspase-9 and caspase-3 activation, and PARP cleavage.
In our study, overexpression of Bcl-2 could suppress the apoptotic effects of galangin on HCC cells. Bcl-2 is an important anti-apoptotic protein that suppresses different drug-induced activation of the mitochondria-apoptotic pathway, such as etoposide[23], berberine[24], safatoposide[25], epigallocatechin-3-gallate[26], curcumin[27] and anti-inflammatory drugs[28]. The Bcl-2 protein can block the oligomerization of Bax and Bak and inhibit the apoptotic program[29,30]. Moreover, our data also showed that Bcl-2 decrease could enhance HCC cell sensitivity to galangin. These results show that Bcl-2 can modulate the effects of galangin on HCC cells and indicate that galangin induces apoptosis viathe mitochondrial pathway.
We demonstrated that AIF is released from mitochondria into the cytosol in HCC cells upon galangin treatment. AIF migrates into the nucleus and induces high-molecular-mass DNA fragmentation and marginal chromatin condensation independent of caspases[31,32]. Therefore, HCC cells undergoing apoptosis may be due to a combination of caspase activation and AIF release.
In summary, we demonstrate that galangin induces HCC cell apoptosis via the mitochondrial pathway. Our data demonstrated that (1) galangin induces HCC cell apoptosis by triggering Bax translocation to the mitochondria; (2) galangin-treated HCC cells causes the release of AIF and cytochrome c into the cytosol from the mitochondria; and (3) overexpression of Bcl-2 attenuated galangin-induced HepG2 cells apoptosis, while down-regulated Bcl-2 expression enhanced galangin to induce cell apoptosis.

Our observation exhibits that GA treatments upregulated levels of p21 and p27, whereas downregulated levels of CDK4-cyclin D1 and cyclin E-CDK2 complexes. Taken together, it suggests that induction of G1 phase arrest of TNBC cells by GA treatments may attribute to upregulation of p21 and p27, and the consequent disrupting CDK4-cyclin D and CDK2-cyclin E complexes. Mounting evidences have indicated that cell-cycle arrest and apoptosis may be linked. CDK inhibitors have been suggested to be indirectly involved in apoptosis through regulation of CDKs. p21 can be promoted by both p53-dependent and p53-independent mechanisms following stress [42]. Moreover, overexpression of p21 has been reported to trigger apoptosis [43]. Our findings show that GA treatment increased ratio of sub-G1 phase and number of apoptotic cells in MDA-MB-231 cells, suggesting that GA treatment not only induced G1 phase arrest but also triggered apoptosis of MDA-MB-231 cells.Stress signals can activate p38 MAPK cascades and subsequently mediate cellular responses such as cellular apoptosis.
Conclusion The present study demonstrates that GA treatment significantly reduces cell viability of malignant human breast cancer cell line MDA-MB-231 via induction of both G1 phase arrest and apoptosis, which may attribute to the increase of p21 and p27 by activation of p53. These findings indicate that GA may provide potent
Gallic Acid Induces Apoptosis in 3T3-L1 Pre-adipocytes via a Fas- and Mitochondrial-Mediated Path
Gallic acid (3,4,5-trihydroxybenzoic acid) is a naturally abundant plant phenolic compound. Our previous studies have shown that some phenolic acids such as gallic acid inhibit cell growth and induce apoptosis in 3T3-L1 pre-adipocytes. However, the molecular mechanism of gallic acid in the induction of cell apoptosis is still unclear. In this study, we investigated the effect of gallic acid on the apoptotic pathway in 3T3-L1 pre-adipocytes. Western blot data revealed that gallic acid stimulated an increase in the protein expression of Fas, FasL, and p53. The ratio of expression levels of pro- and anti-apoptotic Bcl-2 family members was changed by gallic acid treatment. Gallic acid released mitochondrial cytochrome c into the cytosol and subsequently induced the activation of caspase-9 and caspase-3, which were followed by the cleavage of poly(ADP-ribose) polymerase. Pretreatment with a general caspase-9 inhibitor (Z-LEHD-FMK) and caspase-3 inhibitor (Z-DEVD-FMK) prevented gallic acid from inhibiting cell viability in 3T3-L1 pre-adipocytes. The data also indicated that treatment with gallic acid inhibited histone deacetylase activity in 3T3-L1 pre-adipocytes. These results demonstrate that gallic acid induces apoptosis in 3T3-L1 pre-adipocytes through the Fas and mitochondrial pathway. The induction of cell apoptosis by gallic acid may prove to be a pivotal mechanism for decreased pre-adipocyte proliferation.
Keywords: Gallic acid; apoptosis; 3T3-L1 pre-adipocytes

Gambogic Acid
(Garcinia hanburyi Hook. aka “Hanbury’s garcinia”)
matrix metalloproteinase (MMP) 2 and 9 suppression
Cancer cell invasion is one of the crucial events in local spreading, growth, and metastasis of tumors. The present study investigated the antiinvasive and antimetastatic action of gambogic acid (GA) in MDA-MB-435 human breast carcinoma cells. GA caused a concentration-dependent suppression of cell invasion through Matrigel and significantly inhibited lung metastases of the cells transplanted in vivo. The potent effects of GA have been attributed to its ability to reduce the expression of matrix metalloproteinases (MMP) 2 and 9 in vitro and in vivo both at the protein and mRNA levels, which were associated with protein kinase C (PKC) signaling pathway as supported by the diminished antiinvasive effect of GA in the presence of specific activator of the pathway. Collectively, our data demonstrated that GA exhibited antiinvasion properties on highly invasive cancer cells via PKC mediated MMP-2/9 expression inhibition. This indicated that GA can be served as a potential novel therapeutic candidate for the treatment of cancer metastasis.
Gambogic acid, a potent inhibitor of survivin, reverses docetaxel resistance in gastric cancer cells.
OBJECTIVE: Chemoresistance is a major obstacle to successful cancer chemotherapy. In this study, we examined the ability of gambogic acid (GA) to reverse docetaxel resistance in BGC-823/Doc gastric cancer cells.
METHODS: The cytotoxic and apoptotic effect of drugs were evaluated by MTT assay and double staining with both Annexin-V-FITC and PI. Cell cycle analysis was determined by PI-stained flow cytometry. Expression of survivin and bcl-2 were evaluated by real-time quantitative RT-PCR.
RESULTS: Treatment of BGC-823/Doc cells with gambogic acid at concentrations of 0.05 microM, 0.1 microM, and 0.2 microM, led to a dramatic increase in docetaxel-induced cytotoxicity without any cytotoxicity by itself. In parallel, gambogic acid treatment caused an increase in apoptotic cell death by docetaxel. Cell cycle analysis indicated that gambogic acid treatment potentiated docetaxel-induced G2/M arrest. Analysis of apoptotic associated gene revealed that gambogic acid singly or in combination with docetaxel significantly downregulate the mRNA expression of survivin, while with no effect on bcl-2.
CONCLUSIONS: Our results describe the potential role of gambogic acid to reverse docetaxel resistance though downregulation of survivin, which may make it an attractive new agent for the chemosensitization of cancer cells.

Ganoderma lucidum (Reishi)
Ganoderma Lucidum (Reishi) Inhibits Cancer Cell Growth and Expression of Key Molecules in Inflammatory Breast Cancer

Genistein
(from Lycium barbarum L. aka “Chinese wolfberry” and “Himalayan /Tibetan goji”)
inhibits tyrosine kinase
Genistein, a natural isoflavonoid phytoestrogen, is a strong inhibitor of protein tyrosine kinases. We analyzed the effects of genistein on in vitro growth, cell-cycle progression and chromatin structure of Jurkat cells, a T-cell leukemia line with a constitutively increased tyrosine phosphorylation pattern. Exposure of in vitrocultured Jurkat cells to genistein resulted in a dose-dependent, growth inhibition. Cell-cycle analysis of genistein-treated cells revealed a G2/M arrest at low genistein concentrations (5–10 μg/ml), while at higher doses (20–30 μg/ml) there was also a perturbation in S-phase progression. The derangements in cell-cycle control were followed by apoptosic death of genistein-treated cells. Immunocytochemical analysis of cells stained with a FITC-conjugated anti-phosphotyrosine monoclonal antibody showed that 30 μ/ml genistein effectively inhibit tyrosine kinase activityin cultured Jurkat cells. Our results indicate that the natural isoflavone genistein antagonizes tumor cell growth through both cell-cycle arrest and induction of apoptosis and suggest that it could be a promising new agent in cancer therapy.
Inhibits BCL-2
Genistein inactivates bcl-2, delays the G2/M phase of the cell cycle, and induces apoptosis of human breast adenocarcinoma MCF-7 cells.The aim of this study was to identify the molecular mechanism of action of the isoflavone, genistein. Genistein at 0.15 mM caused MCF-7 apoptotic cell death, which was accompanied by cell cycle delay in the G2/M phase. Twenty-four hours post-treatment, 47.3% of the MCF-7 cells accumulated at G2/M, compared with 19.9% in the untreated controls. At 0.15 mM, genistein caused an increase in the steady-state levels of the wild-type tumour suppressor p53, which was attributed to stabilising the tumour suppressor protein, since p53 mRNA levels did not increase. Prior to the upregulation of p53, which became evident within 6 h of genistein treatment, there was increased bcl-2 phosphorylation at 30 min post-treatment. Although early changes (30-120 min) in the phosphotyrosine peptide patterns were not detected, after 24h, genistein inhibited phosphorylation of several peptides. These results suggest that genistein’s dual roles of protein tyrosine kinase inhibitor and topoisomerase II inhibitor are essential for the initiation of apoptosis.
Hsp90 INHIBITIOR
Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function.Androgen receptor (AR) is a ligand-activated transcription factor belonging to the steroid hormone receptor family and is very important for the development and progression of prostate cancer. The soy isoflavone genistein has been shown previously to down-regulate AR in androgen-dependent prostate cancer cell lines such as LNCaP. However, the mechanism(s) by which AR is down-regulated by genistein is still not known fully. We show a new mechanism by which genistein inhibits AR protein levels. We show that genistein-treated LNCaP cells exhibit increased ubiquitination of AR, suggesting that AR protein is down-regulated via a proteasome-mediated pathway. AR is normally stabilized by the chaperone activity of the heat shock protein Hsp90. The increased ubiquitination of AR after genistein treatment is attributed to decreased Hsp90 chaperone activity as assessed by its increased functionally inactive acetylated form. Consistent with this result, we find that HDAC6, which is a Hsp90 deacetylase, is inhibited by the antiestrogenic activity of genistein. Hence, in this study, we elucidate a novel mechanism of AR down-regulation by genistein through inhibition of HDAC6-Hsp90 cochaperone function required to stabilize AR protein. Our results suggest that genistein could be used as a potential chemopreventive agent for prostate cancers along with known inhibitors of HDAC6 and Hsp90.
Bcl-2, Bcl-XL & CDC2 downregulation
Genistein, biochanin-A, and daidzein, the predominant soy isoflavones, have been reported to lower the risk of cancer, but it is not known whether they protect against human hepatoma cancer. This study was designed to investigate their effects on cell growth, the cell cycle, and apoptosis induction in the human hepatoma cell lines, HepG2, Hep3B, Huh7, PLC, and HA22T. Genistein, biochanin-A, and daidzein inhibited growth of all five lines in a dose-dependent manner. DNA fragmentation studies and the TUNEL assay demonstrated that isoflavones caused tumor cell death by induction of apoptosis. Activation of caspase-3 and cleavage of the caspase-3 substrate, poly(ADP-ribose)polymerase, was seen in hepatoma cells after 24 hours’ exposure to isoflavones. In addition, isoflavone cytotoxicity correlated with downregulation of Bcl-2 and Bcl-XL expression. Synergistic effects of the three isoflavones were observed on cell growth inhibition, apoptosis induction, and anti-apoptotic protein expression. Flow cytometry showed that genistein, but not biochanin-A or daidzein, induced progressive and sustained accumulation of hepatoma cancer cells in the G2/M phase as a result of inhibition of Cdc2 kinase activity. Coapplication of caffeine prevented this cell cycle arrest, but not apoptosis, showing that cell cycle arrest was not necessary for apoptosis. Furthermore, the isoflavones combination also had a significant tumor-suppressive effect in nude mice. These results suggest that isoflavones might be promising agents for the treatment of human hepatoma.
Inhibits HIF-1a & VEGF
Antiangiogenic activity of genistein in pancreatic carcinoma cells is mediated by the inhibition of hypoxia‐inducible factor‐1 and the down‐regulation of VEGF gene expression
Previous reports indicate that Genistein, a naturally occurring isoflavonoid, exhibits strong antiangiogenic activity. The underlying mechanism of inhibition, however, remains unclear. Among the biologic effects of Genistein are the inhibition of tyrosine kinases and the inhibition of hypoxic activation of hypoxia‐inducible factor‐1 (HIF‐1), one of the main regulators of VEGF gene expression.
METHODS: Hypoxic cell culture was performed in a modular incubator chamber. Vascular endothelial growth factor (VEGF) protein secretion was measured using the enzyme‐linked immunosorbent assay, binding of DNA by HIF‐1 was measured using the electrophoretic mobility shift assay, and mRNA quantification was performed using Northern blot analysis. Pancreatic carcinoma was studied in an orthotopic murine model. Angiogenesis in vivo was quantified by staining xenograft tumors for endothelial cell markers.
RESULTS: VEGF protein secretion was dose‐dependently suppressed with increasing doses of Genistein. Furthermore, treatment of pancreatic carcinoma cells with Genistein led to impaired activation of HIF‐1 under hypoxic culture conditions. Northern blot analysis indicated that VEGF mRNA expression decreased upon treatment with Genistein, both under normoxic and hypoxic culture conditions. In vivo, Genistein inhibited tumor growth for xenograft pancreatic carcinoma cells, whereas extensive hypoxia was observed in xenograft tumors and was not influenced by Genistein therapy. Similarly, decreased VEGF mRNA levels were observed in Genistein‐treated Capan‐1 xenograft tumors.
CONCLUSIONS: The current study indicates that the previously reported antiangiogenic activity of Genistein probably is mediated by the inhibition of HIF‐1, an important regulator ofVEGF gene homeostasis, particularly under low‐oxygen conditions. Therefore, this bioactive compound may well be beneficial to patients with pancreatic carcinoma.
genistein inhibits CDK2 & CDC2
Following genistein treatment of cells, an increased binding of p21 with Cdk2 and Cdc2 paralleled a significant decrease in Cdc2 and Cdk2kinase activity with no change in Cdk2 and Cdc2 expression. Genistein also induced the activation of a p21 promoter reporter construct, utilizing a sequence distinct from the p53-binding site. Analysis of deletion constructs of the p21 promoter indicated that the response to genistein could be localized to the 300 base pairs proximal to the transcription start site. These data suggest that genistein may exert a strong anticarcinogenic effect, and that this effect possibly involves an induction of p21, which inhibits the threshold kinase activities of Cdks and associated cyclins, leading to a G2/M arrest in the cell cycle progression.
Ginsenoside Rg3
(a ginsenoside found in Panax ginseng and Panax japonicus var. major)
key function: Rg3 greatly suppresses the major components of SASP, IL-6 & IL-8.
Inhibition of NF-kappaB by ginsenoside Rg3
Ginsenoside Rg3, the main constituent isolated from Panax ginseng, has been of interest for use as a cancer preventive or therapeutic agent. We investigated here whether Rg3 can inhibit the activity of NF-kappaB, a key transcriptional factor constitutively activated in colon cancer that confers cancer cell resistance to chemotherapeutic agents. To investigate whether RG3 can suppress activation of NF-kappaB, and thus inhibit cancer cell growth, we examined the susceptibility of colon cancer cells (SW620 and HCT116) to treatment with Rg3 (25, 50, 75, 100 microM) and RG3-induced activation of NF-kappaB. RG3 dose-dependently inhibited cancer cell growth through induction of apoptosis and decreased NF-kappaB activity. In a further study of compounds in colon cancer, we used half of the IC(50) dose, values in combined treatments of Rg3 (50 microM) with conventional agents – docetaxel (5 nM), paclitaxel (10 nM) cisplatin (10 microM) and doxorubicin (2 microM). Compared to treatment with Rg3 or chemotherapy alone, combined treatment was more effective (i.e., there were synergistic effects) in the inhibition of cancer cell growth and induction of apoptosis and these effects were accompanied by significant inhibition of NF-kappaB activity. NF-kappaB target gene expression of apoptotic cell death proteins (Bax, caspase-3, caspase-9) was significantly enhanced, but the expression of anti-apoptotic genes and cell proliferation marker genes (Bcl-2, inhibitor of apoptosis protein (IAP-1) and X chromosome IAP (XIAP), Cox-2, c-Fos, c-Jun and cyclin D1) was significantly inhibited by the combined treatment compared to Rg3 or docetaxel alone. These results indicate that ginsenoside Rg3 inhibits NF-kappaB, and enhances the susceptibility of colon cancer cells to docetaxel and other chemotherapeutics. Thus, ginsenoside Rg3 could be useful as an anti-cancer or adjuvant anti-cancer agent.
Some saponins, like ginsenosides, have been investigated for their anti-metastatic activity in human breast cancer (Nag et al., 2012). It has also been suggested that certain ginsenosides can prevent cartilage collagen matrix breakdown in patients with arthritis (Lee et al., 2014). Testing for their anti-senescence properties revealed that ginsenoside Rb1 was able to reverse the unfavorable effects of H2O2 treatment in HUVECs through a reduction in malondialdehyde concentrations, an increase in superoxide dismutase activity, a reduction in SA β-gal ac- tivity, and induction of SIRT1 expression (Liu et al., 2011; Song et al., 2014). It also restored normal conditions in human WI-38 diploid fi- broblasts after induction of premature senescence with tert-butyl hy- droperoxide (t-BHP). Senescent fibroblasts typically showed elevated p21 and p16 levels and reduced ATP synthesis associated with cell cycle arrest. Treatment with ginsenoside Rg1 attenuated these features and restarted the cell cycle towards the S phase, thereby delaying senes- cence (Chen et al., 2008). In a study involving the D-galactose-induced mouse aging model, ginsenoside Rg1 also exerted neuroprotective ef- fects, since it i) protected hippocampal stem cells by raising SOX2 levels and glutathione peroxidase and superoxide dismutase activity, thus enhancing telomerase activity and promoting telomere elongation, and ii) reduced inflammation by reducing IL-1β, IL-6 and TNF-α levels and downregulating p53, p21Cip1/Waf1 and p19Arf gene expression, ultimately inducing a general improvement in cognitive ability and neurogenesis (Zhu et al., 2014). Two similar studies of D-galactose-induced mouse pancreas (Dong et al., 2017) and kidney (Fan et al., 2016) aging de- monstrated that ginsenoside Rg1 ameliorated aging-related conditions, reducing senescence markers and the number of damaged cells.
Anti-cancer natural products isolated from chinese medicinal herbs
Extracted from Panax ginseng C.A. Mey. (Renshen) and Panax quinquefolius L., Araliaceae (Xiyangshen), ginsenoside Rg3 (Figure 1N) is a biologically active component with both in vitro and in vivo anti-cancer activities [227, 228]. The anti-proliferative mechanism of ginsenoside Rg3 is associated with the inactivation of NF-κB [229, 230], modulation of MAPKs [231] and the down-regulation of Wnt/β-catenin signaling [232]. Ginsenoside Rg3 affects the ephrin receptor pathway in HCT-116 human colorectal cancer cells [233]. The anti-proliferative mechanism of ginsenoside Rg3 is also associated with the molecules of mitotic inhibition, DNA replication, repair, and growth factor signaling [234].
Ginsenoside Rg3 inhibits the proliferation of HUVEC and suppresses the capillary tube formation of HUVEC on a matrigel at nanomole scales in the presence or absence of VEGF. Ginsenoside Rg3 attenuates VEGF-induced chemo-invasion of HUVEC and ex vivo microvascular sprouting in rat aortic ring. bFGF-induced angiogenesis may be abolished by ginsenoside Rg3[227]. In lung metastasis models of ovarian cancer, ginsenoside Rg3 decreases the number of tumor colonies in the lung and vessels oriented toward the tumor mass [235]. This effect may be partially due to the inhibition of angiogenesis and the decrease in MMP9 expression [235].
Ginsenoside Rg3 increases the efficacy of cancer chemotherapy. Combined treatments with ginsenoside Rg3 enhance the susceptibility of colon cancer cells to docetaxel, paclitaxel, cisplatin and doxorubicin; the mechanism of such an enhancement is related to the inhibition of the constitutively activated NF-κB [229]. A similar phenomenon has been observed in prostate cancer cells, in which the combination of ginsenoside Rg3 and docetaxel more effectively induces apoptosis and G1 cell cycle arrest, accompanied by the inhibition of NF-κB activity [230]. Low-dose administration of cyclophosphamide (CTX) with ginsenoside Rg3 increases the efficacy of targeting the tumor microvasculature and the two-drug combination treatment results demonstrate the longest patient survival rates [236]. Ginsenoside Rg3 combined with gemcitabine not only enhances the efficacy of tumor growth suppression and survival prolongation, but also decreases VEGF expression and microvessel density in tumors [228].
GLYCITEIN
Stat3 NFKB INHIBITOR
Glycitein is an isoflavone that reportedly inhibits the proliferation of human breast cancer and prostate cancer cells. However, its anti‐cancer molecular mechanisms in human gastric cancer remain to be defined. This study evaluated the antitumor effects of glycitein on human gastric cancer cells and investigated the underlying mechanisms. We used MTT assay, flow cytometry and western blotting to investigate its molecular mechanisms with focus on reactive oxygen species (ROS) production. Our results showed that glycitein had significant cytotoxic effects on human gastric cancer cells. Glycitein markedly decreased mitochondrial transmembrane potential (ΔΨm) and increased AGS cells mitochondrial‐related apoptosis, and caused G0/G1 cell cycle arrest by regulating cycle‐related protein. Mechanistically, accompanying ROS, glycitein can activate mitogen‐activated protein kinase (MAPK) and inhibited the signal transducer and activator of transcription 3 (STAT3) and nuclear factor‐kappaB (NF‐κB) signaling pathways. Furthermore, the MAPK signaling pathway regulated the expression levels of STAT3 and NF‐κB upon treatment with MAPK inhibitor and N‐acetyl‐L‐cysteine (NAC). These findings suggested that glycitein induced AGS cell apoptosis and G0/G1 phase cell cycle arrest via ROS‐related MAPK/STAT3/NF‐κB signaling pathways. Thus, glycitein has the potential to a novel targeted therapeutic agent for human gastric cancer.

Gossypol
Bcl-2 and Bcl-xl inhibitor
Induction of apoptosis and antitumor effects of a small molecule inhibitor of Bcl-2 and Bcl-xl, gossypol acetate, in multiple myeloma in vitro and in vivo
Gossypol is a naturally occurring polyphenolic compound extracted from cotton plants. Recent studies revealed that gossypol is a non-peptidic small molecule inhibitor of Bcl-2/Bcl-xl. The aim of the present study was to investigate the induction of apoptosis and antitumor effects of gossypol acetate in multiple myeloma and the possible mechanism(s) of action. Our results showed that gossypol acetate resulted in a dose- and time-dependent inhibition of multiple myeloma cell proliferation, with an IC50 value to both U266 and Wus1 cells at 2.4, 2.2 µM at 48 h after treatment. Gossypol acetate effectively induced the apoptosis of multiple myeloma cells as demonstrated by typical morphological changes, DNA ladder formation and increase in the percentage of cells in subdiploid peak. Furthermore, colorimetric assays showed activation of both caspase-3 and caspase-9. Bcl-2 and Bcl-xl expression was decreased by 86.5±1.2% and 35.9±3.6%, respectively, after treatment with gossypol acetate at 25 µmol/l for 24 h. Preliminary studies in vivo showed that a growth inhibition (T/C) of 30.9% (gossypol acetate 40 mg/kg) was obtained in Balb/C mice bearing Wus1 cells. In addition, there was no body weight loss for the treated group in comparison with the vehicle mice. Our results demonstrated that the potent inhibitor of Bcl-2 and Bcl-xl gossypol acetate had significant antiproliferative and antiapoptotic effects on multiple myeloma cells in vitro and in vivo. Gossypol acetate may represent a promising new anticancer agent with a novel molecular mechanism and warrants further investigation as a single agent, or in combination with other chemotherapeutics, for human multiple myeloma with Bcl-2 overexpression.
In summary, gossypol is a dual inhibitor of MDM2 and VEGFthat disrupts the molecular interaction between MDM2 protein and VEGF mRNA, induces MDM2 self-ubiquitination and degradation, decreases VEGF mRNA stability and protein translation simultaneously, and therefore exerts anti-cancer effects through apoptotic and anti-angiogenesis pathways in human breast cancer in vitro and in vivo, regardless of the p53 status of the cancer cells. We believe development of these MDM2-VEGF inhibitors as potential anticancer drugs for clinical use is worthwhile and represents a novel strategy for improving cancer outcome.
Antiapoptotic members of the Bcl-2 family proteins are overexpressed in prostate cancer and are promising molecular targets for modulating chemoresistance of prostate cancer.(-)-Gossypol, a
natural BH3 mimetic, is a small-molecule inhibitor of Bcl-2/Bcl-xL/Mcl-1 currently
A natural BH3 mimeticinduces autophagy in apoptosis-resistant prostate cancer via modulating Bcl-2–Beclin1 interaction at endoplasmic reticulum A natural BH3-mimetic, small-molecule inhibitor of Bcl-2, (−)-gossypol, shows promise in ongoing phase II and III clinical trials for human prostate cancer. In this study we show that (−)-gossypol preferentially induces autophagy in androgen-independent (AI) prostate cancer cells that have high levels of Bcl-2 and are resistant to apoptosis, both
in vitro and
in vivo, but not in androgen-dependent (AD) cells with low Bcl-2 and sensitive to apoptosis. The Bcl-2 inhibitor induces autophagy through blocking Bcl-2–Beclin1 interaction, together with downregulating Bcl-2, upregulating Beclin1, and activating the autophagic pathway. The (−)-gossypol-induced autophagy is dependent on Beclin1 and Atg5. Our results show for the first time that (−)-gossypol can also interrupt the interactions between Beclin1 and Bcl-2/Bcl-xL at endoplasmic reticulum, thus releasing the BH3-only pro-autophagic protein Beclin1, which in turn triggers the autophagic cascade. Oral administration of (−)-gossypol significantly inhibited the growth of AI prostate cancer xenografts, representing a promising new regimen for the treatment of human hormone-refractory prostate cancer with Bcl-2 overexpression. Our data provide new insights into the mode of cell death induced by Bcl-2 inhibitors, which will facilitate the rational design of clinical trials by selecting patients who are most likely to benefit from the Bcl-2-targeted molecular therapy.
Objective: To determine the effects of guggulsterone (GS), the active substance in guggulipid, on apoptosis, adipogenesis, and lipolysis using 3T3‐L1 cells.
Methods and Procedures: For apoptosis and lipolysis experiments, mature adipocytes were treated with GS isomers. Viability, apoptosis, and caspase 3/7 activation were quantified using MTS, enzyme‐linked immunosorbent assay (ELISA), caspase‐Glo 3/7 activity assay, respectively. The expression of cytochrome c was demonstrated by western blot. Lipolysis was quantified by measuring the release of glycerol. For adipogenesis experiments, postconfluent preadipocytes were incubated with GS isomers for up to 6 days during maturation. Adipogenesis was quantified by measuring lipid content using Nile Red dye. Western blot was also used to demonstrate the adipocyte‐specific transcription factors peroxisome proliferator–activated receptor γ2 (PPARγ2), CCAAT/enhancer binding protein α (C/EBPα), and C/EBPβ.
Results: In mature adipocytes cis‐GS decreased viability, whereas the trans‐GS isomer had little effect. Both isomers caused dose‐dependent increases in apoptosis and cis‐GS was more effective than trans‐GS in inducing apoptosis. cis‐ and trans‐GS also increased caspase‐3 activity and release of cytochrome c from mitochondria. In maturing preadipocytes, both isomers were equally effective in reducing lipid content. The adipocyte‐specific transcription factors PPARγ2, C/EBPα, and C/EBPβ were downregulated after treatment with cis‐GS during the maturation period. Furthermore, cis‐GS increased basal lipolysis of mature adipocytes, but trans‐GS had no effect.
Discussion: These results indicate that GS isomers may exert antiobesity effects by inhibiting differentiation of preadipocytes, and by inducing apoptosis and promoting lipolysis of mature adipocytes. The cis‐GS isomer was more potent than the trans‐GS isomer in inducing apoptosis and lipolysis in mature adipocytes.
Enhanced pro-apoptotic and anti-adipogenic effects of genistein plus guggulsterone in 3T3-L1 adipocytes

GS25 (PANAX NOTOGINSENG)
Therapeutic Potential and Cellular Mechanisms of Panax Notoginseng on Prevention of Aging and Cell Senescence-Associated Diseases
Owing to a dramatic increase in average life expectancy, most countries in the world are rapidly entering an aging society. Therefore, extending health span with pharmacological agents targeting aging-related pathological changes, are now in the spotlight of gerosciences. Panax notoginseng (Burk.) F. H. Chen, a species of the genus Panax, has been called the “Miracle Root for the Preservation of Life,” and has long been used as a Chinese herb with magical medicinal value. Panax notoginseng has been extensively employed in China to treat microcirculatory disturbances, inflammation, trauma, internal and external bleeding due to injury, and as a tonic. In recent years, with the deepening of the research pharmacologically, many new functions have been discovered. This review will introduce its pharmacological function on lifespan extension, anti-vascular aging, anti-brain aging, and anti-cancer properties, aiming to lay the ground for fully elucidating the potential mechanisms of Panax notoginseng’s anti-aging effect to promote its clinical application.
[Mechanisms of delay endothelial cell replicative senescence by extracts from Panax ginseng, Panax notoginseng and Ligusticum chuanxiong].
To explore effect of extracts from Panax ginseng, P. notoginseng and Ligusticum chuanxiong on human umbilical endothelial cells (HUVECs) replicative senescence.HUVECs were induced to aging by generation cultivating to the eighth cells in order to establish a model of endothelial cells replicative senescence. The cultured HUVECs in vitro were divided into 4 groups, the eighth generation cell-senescence untreated group, Vitamin E group, herbal treated high dose and low dose groups. Changes of HUVECs aging were observed by method of SA-beta-gal stained HUVECs and cells cycle were analyzed. Contents of ROS in cells, the levels of anti-superoxide (O2-) and nitric oxide (NO) in cell mediums were examined. Western blot were used to analyse protein expression of NADPH oxidase p47phox, angiotensin type 1 and 2 receptor (AT1R, AT2R).Compared with Vitamin E group, the positive cell numbers of beta-gal stained HUVECs were enhanced, cell proliferation was depressed, and the fluorescence intensity of ROS was increased, at the same time, less NO and more O2- in cells were produced in the eighth generation cell-senescence untreated group. Protein expression of p47phox, AT1R and AT2R in cells increased compared with Vit E group. Chinese herbs of high dose and low dose could improve condutions of HUVECs aging. Chinese herbs of high dose and low dose could reduce the positive cell numbers of beta-gal stained HUVEC, increase cell proliferation and decrease fluorescence intensity of ROS in cells, at the same time, cells secreting more NO and less O2-. Protein expression of p47phox, AT1R and AT2R in cells treated with Chinese herbs of high dose and low dose were decreased compared with Vit E group.The study indicated that extracts from P. ginseng, P. notoginseng and L. chuanxiong could delay endothelial cell replicative senescence. Herbal extracts downregulate the expression of NAD (P) H oxidase subunit-p47phox by means of ROS, hence decrease O2- production and ultimately delay HUVECs in vitro senescence.
[Study on protective effect of Panax notoginseng total saponins on H9c2 cells senescence against D-galactose].
To investigate the protective effect of Panax notoginseng Total Saponins (PNTS) on D-galactose-induced H9c2 cell senescence and the underlying mechanism.D-galactose was used to cause H9c2 cells senescence. Different concentrations of PNTS (5,25 and 50 μg/mL) were added into medium to protect H9c2 cells. Cell senescence was identified by senescence associated β-galactosidase. Level of reactive oxgen species (ROS) was observed according to the effect of DCFH-DA detection. The activity of superoxide dicmutase (SOD) and the content of malondialdehyde (MDA) in cells were measured by biochemical assay kits. The apoptosis of cells was tested by Hochest.Compared with the control group, the number of β-galactosidase positive cells and the fluorescence intensity of ROS in the model group were markedly increased. Meanwhile, the activity of SOD was decreased whereas the content of MDA was increased. The apoptosis level assessed by the Hochest dyeing was significantly increased with chromatin concentration and condensation. Compared with the model group, the quantity of β-galactosidase in different PNTS treatment group was obviously decreased, the activity of SOD was increased and the content of MDA was reduced. The apoptosis rate in the cells treated with PNTS was improved. PNTS improved D-galactose-induced H9c2 cell senescence through upregulation of antioxidative ability and attenuation of cell apoptosis.
[Effect of Panax notoginseng saponins on syp and tau gene expression in brain of senescence accelerated mouse].
OBJECTIVE: To study the effect of Panax notoginseng saponins (PNS) on (synaptophysin, syp) and tau gene expression in the brain tissue in senescence accelerated mouse prone 8 (SAMP 8). METHOD: SAMP8 were randomly divided into 4 groups: PNS 23.38, 93.50 mg x kg(-1) group, huperzin A 0.038 6 mg x kg(-1) x d(-1) group and blank control group; the drug groups were treated with the designed drugs respectively per day by intragastric administration for 4 consecutive weeks, and double distilled water was given to blank control group. After treatment, the mRNA content of tau and syp were assayed by reverse transcription (RT) and real-time polymerase chain reaction (real-time PCR). RESULT: Compared with blank control group, the syp mRNA contents were increased in PNS groups (P < 0.05 or P < 0.01), and the tau mRNA content were not significant difference in all groups. CONCLUSION: This study suggests that PNS can up-regulate syp gene expression at transcriptional level in the brain of SAMP 8.
[Protective effect of total saponins of Panax notoginseng combined with total flavonoids of epimedium on D-galactose-incuced senescence of H9c2 cell].
To investigate the protective effect of Panax notoginseng saponins combined with total flavonoids of epimedium on D-gal-induced senescence of H9c2 cells and explore its underlying mechanisms. The 50 mol•L⁻¹ D-gal was used to induce H9c2 cells senescence. Different concentrations of TPNS, TFE, and TPNS combined with TFE were used for 4 hours for pre-treatment. D-gal was used to stimulate H9c2 cardiac muscle cells for 24 h. Then in order to determine the best combined scheme, MTT was used to detect cell viability. Cell senescence was identified by β-galactosidase staining. Levels of reactive oxygen species(ROS) was observed by DCFH-DA detection. The changes of mitochondrial membrane potential were identified by JC-1 detection. Protein levels of silentmating type information regulation 2 Homolog-1(SIRT1), peroxisomal proliferator-activated receptor-coactivator 1α(PGC-1α) and silentmating type information regulation 2 Homolog-3(SIRT3) were detected by western blot analysis. The results showed that TPNS(5 mg•L⁻¹) combined with TFE(5 mg•L⁻¹) had significant synergistic effect on H9c2 myocardial cell proliferation(Q=1.154), so 5 mg•L-1TPNS combined with 5 mg•L⁻¹ TFE was determined as the best scheme. The quantity of β-galactosidase staining and the fluorescence intensity of ROS were apparently decreased in 5 mg•L⁻¹ TPNS combined with 5 mg•L⁻¹ TFE scheme. Meanwhile, it markedly increased the florescence intensity of mitochondrial membrane potential and enhanced the protein expression of SIRT1, PGC-1α and SIRT3. TPNS combined with TFE could protect H9c2 cells from D-gal-induced senescence.The mechanism might be related to adjusting the signal pathways of SIRT1/PGC-1α, SIRT3, adjusting the structure and function of mitochondria and reducing oxidative stress injury.
anticancer ginsenoside 25-OCH3-PPD, a natural inhibitor of the MDM2 oncogene: Nanoparticle preparation, characterization, in vitro and in vivo anti-prostate cancer activity, and mechanisms of action
The Mouse Double Minute 2 (MDM2) oncogene plays a critical role in cancer development and progression through p53-dependent and p53-independent mechanisms. Both natural and synthetic MDM2 inhibitors have been shown anticancer activity against several human cancers. We have recently identified a novel ginsenoside, 25-OCH3-PPD (GS25), one of the most active anticancer ginsenosides discovered thus far, and have demonstrated its MDM2 inhibition and anticancer activity in various human cancer models, including prostate cancer. However, the oral bioavailability of GS25 is limited, which hampers its further development as an oral anticancer agent. The present study was designed to develop a novel nanoparticle formulation for oral delivery of GS25. After GS25 was successfully encapsulated into PEG-PLGA nanoparticles (GS25NP) and its physicochemical properties were characterized, the efficiency of MDM2 targeting, anticancer efficacy, pharmacokinetics, and safety were evaluated in in vitro and in vivo models of human prostate cancer. Our results indicated that, compared with the unencapsulated GS25, GS25NP demonstrated better MDM2 inhibition, improved oral bioavailability and enhanced in vitro and in vivo activities. In conclusion, the validated nano-formulation for GS25 oral delivery improves its molecular targeting, oral bioavailability and anticancer efficacy, providing a basis for further development of GS25 as a novel agent for cancer therapy and prevention.

HESPERIDIN
NF-KB INHIBITOR
Gout arthritis is a painful inflammatory disease induced by monosodium urate (MSU) crystals. We evaluate the therapeutic potential of the flavonoid hesperidin methylchalcone (HMC) in a mouse model of gout arthritis induced by intra-articular injection of MSU (100 μg/10 μL). Orally given HMC (3-30 mg/kg, 100 μL) reduced in a dose-dependent manner the MSU-induced hyperalgesia (44%, p < 0.05), edema (54%, p < 0.05), and leukocyte infiltration (70%, p < 0.05). HMC (30 mg/kg) inhibited MSU-induced infiltration of LysM-eGFP+cells (81%, p < 0.05), synovitis (76%, p < 0.05), and oxidative stress (increased GSH, FRAP, and ABTS by 62, 78, and 73%, respectively; reduced O2– and NO by 89 and 48%, p < 0.05) and modulated cytokine production (reduced IL-1β, TNF-α, IL-6, and IL-10 by 35, 72, 37, and 46%, respectively, and increased TGF-β by 90%, p < 0.05). HMC also inhibited MSU-induced NF-κB activation (41%, p < 0.05), gp91phox (66%, p < 0.05) and NLRP3 inflammasome components mRNA expression in vivo (72, 77, 71, and 73% for NLRP3, ASC, pro-caspase-1, and pro-IL-1 β, respectively, p < 0.05), and induced Nrf2/HO-1 mRNA expression (3.9- and 5.1-fold increase, respectively, p < 0.05). HMC (30, 100, and 300 μM) did not inhibit IL-1β secretion by macrophages primed by LPS and challenged with MSU (450 μg/mL), demonstrating that the anti-inflammatory effect of HMC in gout arthritis depends on inhibiting NF-κB but not on direct inhibition of inflammasome.The pharmacological effects of HMC indicate its therapeutic potential for the treatment of gout.
UPREGULATES NRF2
KEY FINDINGS: Hesperidin treatment effectively protected aged rat heart by increasing the activity of enzymic antioxidants. Hesperidin upregulated the protein levels of nuclear factor erythroid 2-related factor 2, which is responsible for maintaining the antioxidant status of the cell.
CONCLUSIONS: Hesperidin could be useful in protecting cardiomyocytes against age-related increase in oxidative stress mediated by Nrf2 upregulation.
ANTI SENESCENCE EFFECTS
Citrus unshiu peel extract, containing hesperetin (metabolite of hesperidin), decreased expression levels of 𝛽-galactosidase, matrix metalloproteinase-1, and the number of senescent cells. Moreover, pretreatment of human fibroblasts with hesperetin glucuronides induced a 25% protection against UV-A-induced necrotic cell death [38].
In addition to upregulation of Nrf2 expression in the senescent rat heart [115], methylhesperidin, methylated derivative of hesperidin, enhanced translocation of Nrf2 from cytoplasm to nuclear, resulting in upregulation of antioxidant- related gene expression and reduction in reactive oxygen species, consequently leading to protection of epidermal keratinocytes against UVB-induced damage in keratinocyte cultures [116].
P38 MAPK INHIBITOR
AttenuationofInflammation. Developmentofinflamma- tion is a complex process involving interactions of a number of molecules in various signaling pathways, including p38 mitogen-activated protein kinase (MAPK) pathway [133]. Inhibition of p38 MAPK signaling pathway can markedly lower expression of IL-1𝛽 and IL-6, IL-8, IL-18, and TNF𝛼, in both macrophage culture and mice [134, 135]. Study showed that, prior to H2O2 stimulation, treatment of keratinocytes with hesperidin for 2 hr induced over 50% reduction in NF-𝜅B and phosphorylated p38 MAPK in comparison with those without pretreatment with hesperidin [62]. Likewise, treatment of mouse RAW 264.7 cells with hesperetin metabo- lite almost completely reversed lipopolysaccharide-induced increase in NF-𝜅B expression in addition to reductions in phosphorylated p38 MAPK and c-Jun N-terminal kinase 1/2 [61]. Thus, hesperidin-induced inhibition of p38 MAPK signaling pathway could contribute its attenuation of inflammation.
Citrus seeds are full of phenolic compounds, such as flavonoids. The aims of this study were to identify the types of flavonoids in Citrus seed extracts, the cytotoxic effect, mode of cell death, and signaling pathway in human hepatic cancer HepG2 cells. The flavonoids contain anticancer, free radical scavenging, and antioxidant activities. Neohesperidin, hesperidin, and naringin, active flavanone glycosides, were identified in Citrus seed extract. The cytotoxic effect of three compounds was in a dose-dependent manner, and IC50 levels were determined. The sensitivity of human HepG2 cells was as follows: hesperidin > naringin > neohesperidin > naringenin. Hesperidin induced HepG2 cells to undergo apoptosis in a dose-dependent manner as evidenced by the externalization of phosphatidylserine and determined by annexin V-fluorescein isothiocyanate and propidium iodide staining using flow cytometry. Hesperidin did not induce the generation of reactive oxygen species, which was determined by using 2′,7′-dichlorohydrofluorescein diacetate and flow cytometry method. The number of hesperidin-treated HepG2 cells with the loss of mitochondrial transmembrane potential increased concentration dependently, using 3,3′-dihexyloxacarbocyanine iodide employing flow cytometry. Caspase-9, -8, and -3 activities were activated and increased in hesperidin-treated HepG2 cells. Bcl-xL protein was downregulated whereas Bax, Bak, and tBid protein levels were upregulated after treatment with hesperidin in a dose-dependent manner. In conclusion, the bioflavanone from Citrus seeds, hesperidin, induced human HepG2 cell apoptosis via mitochondrial pathway and death receptor pathway. Citrus seed flavonoids are beneficial and can be developed as anticancer drug or food supplement, which still needs further in vivo investigation in animals and human beings.
BLOCKS TLR4
HES decreased the expression of TLR4 protein and increased the expression of GLUT2 protein. Meanwhile, the expression of GLUT2 protein would increased when TLR4 was blocked. Based on the above knowledge, we speculated that HES ameliorated IR by directly affecting the expression of TLR4 and NF‐κB proteins and indirectly affecting the expression of GLUT2 protein by regulating TLR4. However, HES could not affect GLUT2 by TLR4 when TLR4 was blocked. Therefore, the effect of HES on the expression of GLUT2 protein in cells was attenuated compared with the cells that were not treated with HTA125, which reveals that TLR4 was an underlying key target for HES in ameliorating IR.
Honokiol
CDK1⊥, Bcl-2↓, Bax↑, cyclin D1↓, pAKT↓, γ-secretase activity↓, γ-secretase complex proteins↓, PPAR-γ⊥, COX-2⊥, NF-κB⊥, EGFR/P13K/AKt↓; JunB ↓ and JunD↓ caspase-8↑, caspase-9↑, caspase-3↑, PARP↑, p53↑, CD31 staining↓, LH↑, p38⊥, NF-κB⊥, Bcl-XL↓, Bad↑, cyclin E↓, (Cdk2 and Cdk4)↓, Cdk↑, p21 and p27↑, NF-κB↓, Bcl-2↓, Mcl-1↓, surcivin↓, VEGF↓, STAT3⊥, HG-induced IL1β⊥, IL18⊥, TNFα⊥, -PGE2⊥, NO⊥, and TGFβ1⊥, MCP-1⊥, MIP-1α⊥, EGFR targeting TKI⊥, Akt⊥ erlotinib⊥, EGFR signaling⊥, MAPK⊥, cyclin D1⊥
Honokiol: a novel natural agent for cancer prevention and therapy
Honokiol ((3’,5-di-(2-propenyl)-1,1’-biphenyl-2,2’-diol) is a bioactive natural product derived from Magnolia spp. Recent studies have demonstrated anti-inflammatory, anti-angiogenic, anti-oxidative and anti-cancer properties of honokiol in vitro and in preclinical models. Honokiol targets multiple signaling pathways including nuclear factor kappa B (NF-κB), signal transducers and activator of transcription 3 (STAT3), epidermal growth factor receptor (EGFR) and mammalian target of rapamycin (m-TOR), which have great relevance during cancer initiation and progression. Furthermore, pharmacokinetic profile of honokiol has revealed a desirable spectrum of bioavailability after intravenous administration in animal models, thus making it a suitable agent for clinical trials. In this review, we discuss recent data describing the molecular targets of honokiol and its anti-cancer activities against various malignancies in pre-clinical models. Evaluation of honokiol in clinical trials will be the next step towards its possible human applications.
INHIBITS STAT3
Honokiol suppressesSTAT3 activity induced by IL-6, one of the many growth factors that activate STAT3(25). Furthermore, STAT3 inhibition by honokiol has also been correlated with the repression of upstream protein tyrosine kinases c-Src, JAK1 and JAK2 (26). Although not yet demonstrated, suppression of STAT3 and NF-κB activation by honokiol could also be inter-linked. The p65 subunit of NF-κB has been shown to interact with STAT3 (27) and it is reported that STAT3 prolongs NF-κB nuclear retention through acetylation (28). Activation of both STAT3 and NF-κB in tumor cells by factors in tumor microenvironment (interleukins and chemokines) and release of cytokines and chemokines by tumor cells as a response (21, 28) also suggest their role in mediating the cross-talk between tumor and its microenvironment. All these observations clearly imply that NF-κB and STAT3 inhibition by honokiol can have a multifaceted impact on the growth and spread of the tumor cells.
PI3k AKT MTOR Inhibitor / Autophagy activator
Polyphenolic compounds such as can honokiol act as anticar cinogenic agents. Honokiol (3-,5-di-(2-propenyl)-1,1′ -biphenyl- 2,2′-diol), a naturally occurring dietary product isolated from an extract of seed cones from Magnolia grandiflora, can induce cellular autophagy by modulating the PI3K/Akt/mTOR signaling pathway in neuroblastoma cells (Yeh et al., 2016). Treatment with honokiol has been shown to reduce PI3K content, and in turn, inhibit Akt, which down regulates phosphorylation of mTOR. mTOR inhibition induces autophagyby upregulating the downstream protein kinases, such as ULK1and ATG13 (Yeh et al., 2016).
DOWNREGULATES BCL-XL
Honokiol is a phenolic compound purified from Magnolia officinalis, which induced the apoptotic cell death in several types of human cancer cells. In the present study, the molecular mechanism of honokiol-mediated apoptotic process was examined in human squamous lung cancer CH27 cells. Here, we found that honokiol-induced apoptotic cell death was accompanied by upregulation of Bad and downregulation of Bcl-XL, while honokiol had no effect on the levels of Bcl-2, Bcl-XS, Bag-1, Bax and Bak proteins. Moreover, honokiol treatment caused the release of mitochondrial cytochrome c to cytosol and sequential activation of caspases. Proteolytic activation of caspase-3 and cleavage of PARP, an in vivo substrate for caspase-3, were observed in honokiol-treated CH27 cells. Furthermore, treatment with caspase inhibitors z-DEVD-fmk and z-VAD-fmk markedly blocked honokiol-induced apoptosis. These results demonstrated that modulation of Bcl-XL and Bad proteins, release of mitochondrial cytochrome c and activation of caspase-3, participated in honokiol-triggered apoptotic process in human squamous lung cancer CH27 cells.
HSP90 INHIBITOR
Here, LHK increased HSP90 acetylation levels , which resulted in inhibition of HSP90 and HCP binding (Fig. 3E). LHK was reported to inhibit class I histone deacetylase [40,41]. The increased HSP90 acetylation levels re- ported here may be due to class I histone deacetylase-mediated inhibition by LHK. Additionally, HK destabilized EGFR proteins in cancer cell lines [40,42]; these findings were recapitulated in the current study. The Akt and Erk1/2 pathways are activated to sustain cell survival in NSCLC cells that have EGFR-activating and genfitinib-resistant mutations. In H1975 and HCC827 cell lines, LHK promoted degradation of EGFR, C-Raf and Akt, and the Akt and Erk1/2 pathways were both inhibited . Taken together, these data indicated that LHK inhibited the Akt and Erk1/2 path- ways by promoting the degradation of HCP in NSCLC cell lines harboring EGFR-activating and resistant mutations.
Honokiol (HNK) is a natural compound isolated from the magnolia plant with numerous pharmacological activities, including inhibiting epithelial-mesenchymal transition (EMT), which has been proposed as an attractive target for anti-tumor drugs to prevent tumor migration. In this study we investigated the effects of HNK on EMT in human NSCLC cells in vitro and the related signaling mechanisms. TNF-α (25 ng/mL) in combination with TGF-β1 (5 ng/mL) was used to stimulate EMT of human NSCLC A549 and H460 cells. Cell proliferation was analyzed using a sulforhodamine B assay. A wound-healing assay and a transwell assay were performed to examine cell motility. Western blotting was used to detect the expression levels of relevant proteins. siRNAs were used to knock down the gene expression of c-FLIP and N-cadherin. Stable overexpression of c-FLIP L (H157-FLIP L) or Lac Z (H157-Lac Z) was also performed. Treatment with TNF-α+TGF-β1 significantly enhanced the migration of A549 and H460 cells, increased c-FLIP, N-cadherin (a mesenchymal marker), snail (a transcriptional modulator) and p-Smad2/3 expression, and decreased IκB levels in the cells; these changes were abrogated by co-treatment with HNK (30 μmol/L). Further studies demonstrated that expression level of c-FLIP was highly correlated with the movement and migration of NSCLC cells, and the downstream effectors of c-FLIP signaling were NF-κB signaling and N-cadherin/snail signaling, while Smad signaling might lie upstream of c-FLIP. HNK inhibits EMT-mediated motility and migration of human NSCLC cells in vitro by targeting c-FLIP, which can be utilized as a promising target for cancer therapy, while HNK may become a potential anti-metastasis drug or lead compound.
Honokiol exerts dual effects on browning and apoptosis of adipocytes
•Honokiol exhibits a dual modulatory role in adipocytes.
•Honokiol regulates lipid catabolism and activates AMPK.
•Honokiol induces brown adipocyte-like phenotype through activation of ERK.
Induction of brown adipocyte-like phenotype (browning) in white adipocytes and promotion of apoptosis by dietary and pharmacological compounds is considered a novel strategy against obesity. Here, we show that honokiol exerts dual modulatory effects on adipocytes via induction of browning in 3T3-L1 white adipocytes and apoptosis as well as activation of HIB1B brown adipocytes combined with inhibition of apoptosis.
Honokiol-induced browning and apoptosis were investigated by determining expression levels of brown adipocyte-specific genes and proteins by RT-PCR and immunoblot analysis, respectively. Apoptotic data were validated by immunofluorescence and ROS levels were measured by FACS analysis.
Honokiol treatment induced browning by elevating expression levels of brown adipocyte-specific genes such as Cidea, Cox8, Fgf21, Pgc-1α, and Ucp1. Honokiol promoted apoptosis of 3T3-L1 white adipocytes and inhibited apoptosis of HIB1B brown adipocytes viaopposite regulation of the pro-apoptotic protein BAX and anti-apoptotic protein Bcl-2. Honokiol also significantly increased protein expression levels of ACOX1, CPT1, p-HSL, and p-PLIN and reduced ROS levels, suggesting its possible role in fat oxidation and lipid catabolism. Honokiol-induced browning could be mediated by activation of ERK, as inhibition of ERK by FR180204 abolished expression of PGC-1α and UCP1.
Our findings suggest that honokiol exhibits a modulatory role in adipocytes via induction of browning and apoptosis in white adipocytes, promotion of catabolic lipid metabolism, as well as activation and inhibition of apoptosis in HIB1B brown adipocytes, thereby exhibiting therapeutic potential against obesity.

We previously reported that hyperforin, a phloro- glucinol purified from Hypericum perforatum, induces the mitochondrial pathway of caspase-dependent apoptosis in chronic lymphocytic leukemia (CLL) cells ex vivo, and that this effect is associated with upregulation of Noxa, a BH3-only protein of the Bcl-2 family. Here, we investigated the role of this upregulation in the pro-apoptotic activity of hyperforin in the cells of CLL patients and MEC-1 cell line. We found that the increase in Noxa expression is a time- and concentration- dependent effect of hyperforin occurring without change in Noxa mRNA levels. A post-translational regulation is suggested by the capacity of hyperforin to inhibit proteasome activity in CLL cells. Noxa silencing by siRNA reduces partially hyperforin-elicited apoptosis. Furthermore, treatment with hyperforin, which has no effect on the expression of the prosurvival protein Mcl-1, induces the interaction of Noxa with Mcl-1 and the dissociation of Mcl-1/ Bak complex, revealing that upregulated Noxa displaces the proapoptotic protein Bak from Mcl-1. This effect is accompanied with Bak activation, known to allow the release of apoptogenic factors from mitochondria. Our data indicate that Noxa upregulation is one of the mechanisms by which hyperforin triggers CLL cell apoptosis.They also favor that new agents capable of mimicking specifically the BH3-only protein Noxa should be developed for apoptosis-based therapeutic strategy in CLL.
Cdc2↓, cyclin B1↓, complex⊥, caspase-9↑, MMP↓, p53↑, Bax↑, COX-2↑, MMP-9↑, TPA⊥, protein E6 and E7⊥, p53⊥, Bax↑, Bcl-2↓, caspase-3↑, p53↑, p21↑, ERK1/2⊥
CASPASE 3 & 9 ACTIVATOR : Induces Apoptosis through Mitochondrial Pathway
We examined the antiproliferation effect of Jaceosidin (4′, 5, 7-trihydroxy-3′, 6-dimethoxyflavone) isolated from the herb of Artemisia vestita Wall on several human cancer cell lines. Jaceosidin significantly reduced the proliferation of CAOV-3, SKOV- 3, HeLa, and PC3 cells in a concentration-dependent manner. A time-dependent inhibition was also observed in CAOV-3 cells by Jaceosidin. By flow cytometric analysis, we found that Jaceosidin treatment resulted in an increased apoptosis in CAOV-3 cells. The cells treated with Jaceosidin exhibited a decreased mitochondrial membrane potential. Jaceosidin also increased the level of cleaved caspase-9 and induced the cleavage of caspase-3 and poly (ADP-ribose) polymerase (PARP), while caspase-3 inhibitor Z-DEVD-FMK significantly reversed the proapoptotic effect of Jaceosidin in CAOV-3 cells. Moreover, Jaceosidin elevated the level of cytochrome c in cytosol. These findings suggest that the anticancer effect of Jaceosidin may be contributed by an induction of apoptosis involving cytochrome c release from mitochondria to cytosol.
DOWNREGULATES CYCLIN B1, CDK1; UPREGULATES P53 AND P21
Flavonoid compounds have been shown to trigger cell cycle arrest at G0/G1, S and G2/M checkpoints, allowing cells to repair DNA damage before entry into mitosis. Jaceosidin, a flavonoid compound, has been reported to induce apoptosis in various cancer cell lines. In our previous study, we established that jaceosidin induces apoptosis in U87 glioblastoma cells through G2/M phase arrest. However the molecular mechanisms oremained unclear. In the present study, mRNA and protein expression levels of major cell cycle regulatory genes were analyzed by semi-quantitative RT-PCR and Western blot studies respectively. The results demonstrated that jaceosidin-induced G2/M phase arrest in U87 cells is associated with DNA fragmentation, up-regulation of p53 and p21and subsequent down-regulation of cyclin B1 and CDK1 expression at mRNA as well as at protein level. These findings provide insights into jaceosidin-induced G2/M phase arrest in U87 glioblastoma cells.
Juglanin
inhibits BCL-2, BCL-XL; enhances BAX, BAD
Juglanin (Jug) is obtained from the crude extract of Polygonum aviculare, exerting suppressive activity against cancer cell progression in vitro and in vivo. Juglanin administration causes apoptosis and reactive oxygen species (ROS) in different types of cells through regulating various signaling pathways. In our study, the effects of juglanin on non-small cell lung cancer were investigated. A significant role of juglanin in suppressing lung cancer growth was observed. Juglanin promoted apoptosis in lung cancer cells through increasing Caspase-3 and poly ADP-ribose polymerase (PARP) cleavage, which is regulated by TNF-related apoptosis-inducing ligand/Death receptors (TRAIL/DRs) relied on p53 activation. Anti-apoptotic members Bcl-2 and Bcl-xl were reduced, and pro-apoptotic members Bax and Bad were enhanced in cells and animals receiving juglanin. Additionally, nuclear factor-κB (NF-κB), phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) and mitogen-activated protein kinases (MAPKs) activation were inhibited by juglanin. Further, juglanin improved ROS and induced autophagy. ROS inhibitor N-acetyl-l-cysteine (NAC) reversed apoptosis induced by juglanin in cancer cells. The formation of autophagic vacoules and LC3/autophagy gene7 (ATG7)/Beclin1 (ATG6) over-expression were observed in juglanin-treated cells. Also, juglanin administration to mouse xenograft models inhibited lung cancer progression. Our study demonstrated that juglanin could be a promising candidate against human lung cancer progression.
Icariin
Icariin (from Epimedium aka “Horny Goat Weed”)
strongly inhibits STAT3 activation
Signal transducer and activator of transcription-3 (STAT3) is critical for cancer progression by regulating tumor cell survival, proliferation, and angiogenesis. Herein, we investigated the regulation of STAT3 activation and the therapeutic effects of Icaritin, a prenyl flavonoid derivative from Epimedium Genus, in renal cell carcinoma (RCC). Icaritin showed significant anti-tumor activity in the human and mouse RCC cell lines, 786-O and Renca, respectively. Icaritin inhibited both constitutive and IL-6-induced phospho-STAT3 (STAT3(Y705)) and reduced the level of STAT3-regulated proteins Bcl-xL, Mcl-1, Survivin, and CyclinD1 in a dose-dependent manner. Icaritin also inhibited activation of Janus-activated kinase-2 (JAK2), while it showed minimal effects on the activation of other key signaling pathways, including AKT and MAPK. Expression of the constitutively active form of STAT3 blocked Icaritin-induced apoptosis, while siRNA directed against STAT3 potentiated apoptosis. Finally, Icaritin significantly blunted RCC tumor growth in vivo, reduced STAT3 activation, and inhibited Bcl-xL and Cyclin E, as well as VEGF expression in tumors, which was associated with reduced tumor angiogenesis. Overall, these results suggest thatIcaritin strongly inhibits STAT3 activation and is a potentially effective therapeutic option for the treatment of renal cell carcinoma.
Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade
Because of the essential role of signal transducer and activator of transcription 3 (STAT3) in proliferation, anti-apoptosis, and chemoresistance of multiple myeloma (MM), we investigated whether icariin, a prenylated flavonol glycoside, inhibits both constitutive and inducible STAT3 activation in human myeloma cell lines. We noted that icariin could block constitutive STAT3 phosphorylation as well as its nuclear translocation and DNA binding ability in U266 cells. Icariin also suppressed IL-6-induced STAT3 activation through the inhibition of upstream kinases (Janus activated kinase-1 and -2, and c-Src). We found that icariin downregulated the protein expression of STAT3 downstream target gene products such as Bcl-2, Bcl-xl, survivin, IAP-1/2, COX-2, VEGF, and matrix metallopeptidase 9 (MMP-9) in a concentration-dependent manner. Moreover, this flavonoid also exhibited the capacity to significantly induce apoptosis and suppress proliferation of MM cells. Interestingly, this agent also significantly potentiated the apoptotic effects of bortezomib through the suppression of STAT3 activation in MM cells. Altogether, our data indicates that the potential application of icariin as a STAT3 blocker in myeloma therapy.
icariin and icaritin ameliorated hippocampus neuroinflammation via inhibiting HMGB1-related pro-inflammatory signals in lipopolysaccharide-induced inflammation …
Highlights
•HMGB1-RAGE signaling and TLR4-XBP1s-NF-κB signaling were involved in neuroinflammation.
•Icariin and icaritin could ameliorate neuroinflammation via suppressing HMGB1-RAGE signaling.
•Icariin and icaritin might promote neuroregeneration via activating TLR4–NF-κB signaling.
Inflammation is a defensive response of the body and is at the center of many diseases’ process like depression. High mobility group protein box 1 (HMGB1), has been proved to function as a pro-inflammatory cytokine. We aim to explore the role of HMGB1 played in the neuroinflammation here. In this study, we used LPS to induce an acute inflammatory response, and to measure the anti-neuroinflammation effect of icariin (ICA) and icaritin (ICT). We found that LPS could increase the expression of HMGB1 in serum and hippocampus, along with a high expression of HMGB1 in the cytoplasm and a high expression of RAGE, which could be rescued by ICA and ICT, and ethyl pyruvate (EP) pretreatment showed similar effects here. We speculated that the translocation of HMGB1 from the nucleus to the cytoplasm played an important role in neuroinflammatory process, and HMGB1-RAGE signal was involved in this process. Furthermore, we found that ICA and ICT treatment activated TLR4-XBP1s related NF-κB signal, which we thought was relevant with the neuroprotective effect of ICA and ICT. However, EP pretreatment suppressed TLR4-XBP1s- endoplasmic reticulum stress related NF-κB signal to anti-inflammatory response, which was almost absolutely opposite with ICA and ICT treatment. We speculated that it might be caused by the duration of inflammation. We supposed that ICA and ICT could ameliorate neuroinflammation in hippocampus via suppressing HMGB1-RAGE signaling and might show a neuroprotective effect via activating TLR4-XBP1s related NF-κB signal at the same time, making it possible to act as an anti-neuroinflammatory drugs.
icariin and icaritin ameliorated hippocampus neuroinflammation via mediating HMGB1 expression in social defeat model in mice
Depression is a chronic, severe, and often life-threatening disease accompanied with impaired neurogenesis. Evidence showed that neuroinflammation played a key role in the process of depression. High mobility group protein box 1 (HMGB1) has been proved to function as a pro-inflammatory cytokine. In this study, we used a social defeat (SD) stress to induce inflammatory response, aiming to explore the relationship between HMGB1 and neuroinflammation. We found that the expression of HMGB1 decreased in mice exposure to SD stress, but showed a high expression of cytoplasmic HMGB1 and a high expression of RAGE, which could be rescued by ICA and ICT. So, we speculated that the translocation of HMGB1 from the nucleus to the cytoplasm might play an important role in neuroinflammatory process, and HMGB1-RAGE signaling was involved in this process. Furthermore, we also found that TLR4-XBP1s-ER stress related NF-κB signaling activation was also involved in HMGB1-related neuroinflammation. However, ICA and ICT treatment activated NF-κB signaling, and we also observed the translocation of HMGB1 into the nucleus and the increased number of neurons in mice hippocampus, indicating that the activation of NF-κB signaling might be related to neuroregeneration. Moreover, recombinant human HMGB1 protein (rHMGB1) pretreatment could suppress HMGB1-RAGE signaling and TLR4-XBP1s-ER stress related NF-κB signaling, resulted in a suppressed microglia activation in mice hippocampus. We supposed that ICA and ICT could ameliorate neuroinflammation in hippocampus via suppressing HMGB1-RAGE signaling and show neuroprotective effects via activating TLR4– NF-κB signaling at the same time, resulting in improving depressive behaviors in mice.
HMGB1 is a major pro-inflammatory SASP factor
Epigenetic manipulation through long non-coding RNAs, miRNA modulators, and other oligonucleotides has yielded promising outcomes in experimental settings.162–165 Some of these molecules are currently clinically evaluated in CVD.166 Epigenetic modification through histone modification via bromodomain inhibitors could suppress atherosclerosis,167 senescence,168 and the SASP,169 indicating high potency for such interventions. The non-histone DNA-binding protein high mobility group protein B1 (HMGB1) is a major pro-inflammatory SASP factor, when externalized from the nucleus into the extracellular environment.39 Soluble HMGB1 promoted atherosclerosis40,41 and CVD risk.170 It remains to be seen if HMGB1 nuclear retention ameliorates the epigenetic landscape of senescent cells and provides benefits in atherosclerosis. Another promising strategy is the epigenetic blockade of p66Shc, since pharmacological inhibitors are not currently available. p66Shc is a promoter of oxidative stress70 and is highly expressed in senescent cells68 and atherosclerosis.171Inhibiting p66Shc its key regulator MiR-34a is promising,172 as p66Shc knock-out led to senescence prevention,68 30% animal lifespan extension,173 and strong inhibition of atherogenesis.

INDIRUBIN (INDIGO BLUE EXTRACT)
Inhibits CDK1
The bisindole indirubin has been described, more than 30 years ago, as being clinically active in the treatment of human chronic myelocytic leukaemia. However, the underlying mechanism of action has remained unclear. We have reported previously that indirubin and its analogues are potent and selective inhibitors of cyclin-dependent kinases (CDK). In this study, we investigated the influence of indirubin and derivatives on CDK1/cyclin B kinase in human tumour cells at concentrations known to induce growth inhibition. Cells of the mammary carcinoma cell line MCF-7, synchronized by serum deprivation, after serum repletion stay arrested in the G(1)/G(0)phase of the cell cycle in the presence of 2 microM indirubin-3′-monoxime. At higher drug concentrations (> or = 5 microM) an increase of the cell population in the G(2)/M phase is additionally observed. Cells synchronized in G(2)/M phase by nocodazole remain arrested in the G(2)/M phase after release, in the presence of indirubin-3′-monoxime (> or =5 microM). After 24 h treatment with 10 microM indirubin-3′-monoxime a sub-G(2)peak appears, indicative for the onset of apoptotic cell death. Treatment of MCF-7 cells with growth inhibitory concentrations of indirubin-3′-monoxime induces dose-dependent inhibition of the CDK1 activity in the cell. After 24 h treatment, a strong decrease of the CDK1 protein level along with a reduction of cyclin B in complex with CDK1 is observed. Taken together, the results of this study strongly suggest that inhibition of CDK activity in human tumour cells is a major mechanism by which indirubin derivatives exert their potent antitumour efficacy.
INHIBITS STAT3
Stat3 protein has an important role in oncogenesis and is a promising anticancer target. Indirubin, the active component of a traditional Chinese herbal medicine, has been shown previously to inhibit cyclin-dependent kinases, resulting in cell cycle arrest. Here, we show that the indirubin derivatives E564, E728, and E804 potently block constitutive Stat3 signaling in human breast and prostate cancer cells. In addition, E804 directly inhibits Src kinase activity (IC50 = 0.43 μM) in an in vitro kinase assay. Levels of tyrosyl phosphorylation of c-Src are also reduced in cultured cells 30 min after E804 treatment. Tyrosyl phosphorylation of Stat3, which is known to be phosphorylated by c-Src, was decreased, and constitutive Stat3 DNA binding-activity was suppressed in cells 30 min after E804 treatment. The antiapoptotic proteins Mcl-1 and Survivin, which are encoded in target genes of Stat3, were down-regulated by indirubin derivatives, followed by induction of apoptosis. These results demonstrate that E804 directly blocks the Src-Stat3 signaling pathway, suggesting that the antitumor activity of indirubin compounds is at least partially due to inhibition of this pathway.
indole-3-carbinol (I3C)
downregulation of Bcl-2, Bcl-XL
I3C, also abundant in cruciferous vegetables, induces apoptosis by significant downregulation of Bcl-2, Bcl-XL, IAP, X chromosome-linked IAP, and cellular FLICE inhibitory protein (c-FLIP)in human prostate cancer cells.
MDM2 INHIBITOR
Coimmunoprecipitation analysis revealed that I3C reduced by 4‐fold the level of MDM2protein that associated with p53. The p53–MDM2 interaction and absence of p21 production were restored in cells treated with I3C and the ATM inhibitor wortmannin. Significantly, I3C does not increase the number of 53BP1 foci or H2AX phosphorylation, indicating that ATM is activated independent of DNA double‐strand breaks. Taken together, our results demonstrate that I3C activates ATM signaling through a novel pathway to stimulate p53 phosphorylation and disruption of the p53–MDM2 interaction, which releases p53 to induce the p21 CDK inhibitor and a G1 cell cycle arrest.
Isoalantolactone (Tumuxiang)
induces apoptosis in SGC-7901 cells via mitochondrial and PI3K/Akt signaling pathways.
Isoalantolactone, a sesquiterpene lactone, possesses anti-fungal as well as cytotoxic properties. In this study, the effects of Isoalantolactone on cell viability, cell cycle, and apoptosis were investigated in human gastric adenocarcinoma SGC-7901 cells. The results demonstrated that Isoalantolactone induced morphological changes and decreased cell viability. Subsequently, we found that Isoalantolactone induced G2/M and S phase arrest, which was associated with a decrease in the expression level of cyclin B1. Apoptosis triggered by Isoalantolactone was visualized using propidium iodide (PI) and Annexin V-FITC/PI staining. Isoalantolactone-induced apoptosis of SGC-7901 cells was associated with the dissipation of mitochondrial membrane potential (ΔΨ m) that was due to the down-regulation of Bcl-2 and up-regulation of Baxthat led to the cleavage of caspase-3. Additionally, it was found that Isoalantolactone was involved in the inhibition of phosphorylation of PI3K/Akt. Isoalantolactone-induced cytotoxicity and apoptosis of SGC-7901 cells involve mitochondria-caspase and PI3K/Akt dependent pathways, which gives the rationale for in vivo studies on the utilization of Isoalantolactone as a potential cancer therapeutic compound.
Isoorientin
induces apoptosis through mitochondrial dysfunction and inhibition of PI3K/Akt signaling pathway in HepG2 cancer cells
Isoorientin (ISO) is a flavonoid compound that can be extracted from several plant species, such as Phyllostachys pubescens, Patrinia, and Drosophyllum lusitanicum; however, its biological activity remains poorly understood. The present study investigated the effects and putative mechanism of apoptosis induced by ISO in human hepatoblastoma cancer (HepG2) cells. The results showed that ISO induced cell death in a dose-dependent manner in HepG2 cells, but no toxicity in human liver cells (HL-7702) and buffalo rat liver cells (BRL-3A) treated with ISO at the indicated concentrations. ISO-induced cell death included apoptosis which characterized by the appearance of nuclear shrinkage, the cleavage of poly (ADP-ribose) polymerase (PARP) and DNA fragmentation. ISO significantly (p < 0.01) increased the Bax/Bcl-2 ratio, disrupted the mitochondrial membrane potential (MMP), increased the release of cytochrome c, activated caspase-3, and enhanced intracellular levels of reactive oxygen species (ROS) and nitric oxide (NO). In addition, ISO effectively inhibited the phosphorylation of Akt and increased FoxO4 expression. The PI3K/Akt inhibitor LY294002 enhanced the apoptosis-inducing effect of ISO. However, LY294002 markedly quenched ROS and NO generation and diminished the protein expression of heme peroxidase enzyme (HO-1) and inducible nitric oxide synthase (iNOS). Furthermore, the addition of a ROS inhibitor (N-acetyl cysteine, NAC) or iNOS inhibitor (N-[3-(aminomethyl) benzyl] acetamidine, dihydrochloride, 1400W) significantly diminished the apoptosis induced by ISO and also blocked the phosphorylation of Akt. These results demonstrated for the first time that ISO induces apoptosis in HepG2 cells and indicate that this apoptosis might be mediated through mitochondrial dysfunction and PI3K/Akt signaling pathway, and has no toxicity in normal liver cells, suggesting that ISO may have good potential as a therapeutic and chemopreventive agent for liver cancer.
MAPK signaling pathways regulate mitochondrial-mediated apoptosis induced by isoorientin
Isoorientin (ISO)is a flavonoid compound that can be extracted from several plant species, such as Phyllostachys pubescens, Patrinia, and Drosophyllum lusitanicum. ISO is able to induce apoptosis through mitochondrial dysfunction and inhibition of PI3K/Aktsignaling pathway in HepG2 cells, however, the effects of ISO on MAPK signaling pathways remain unknown. The present study investigated the effects of ISO on this pathway, and the roles of MAPK kinases on mitochondrial-mediated apoptosis in HepG2 cells. The results showed that ISO induced cell death in a dose- and time-dependent manner, and induction apoptosis is main cause for ISO-induced cytotoxicity in HepG2 cells. ISO significantly inhibited the levels of ERK1/2 kinase and increased the expression of JNK and p38 kinases. Furthermore, U0126 (an ERK1/2 inhibitor) significantly enhanced the ISO-induced the Bax/Bcl-2 ratio, the release of cytochrome c to the cytosol fraction, and the levels of cleaved caspase-3. While SP600125 (a JNK inhibitor) and SB203580 (a p38 inhibitor) markedly prevented the expression of these proteins induced by ISO. Furthermore, the ROS inhibitor (NAC) notably promoted the inhibited effect of ISO on the ERK1/2 kinase. NAC also suppressed the p-JNK and p-p38, but failed to reverse the effects of ISO. These results demonstrated for the first time that ISO induces apoptosis in HepG2 cells through inactivating ERK1/2 kinase and activating JNK and p38 kinases, and ROS stimulated by ISO is able to activate the MAPK singaling pathway as the upstream signaling molecules. Initiating event of the mitochondrial-mediated apoptosis induced by ISO is MAPK signals.
The present study showed that ISO induced autophagy, which was correlated with the formation of autophagic vacuoles and the overexpression of Beclin-1 and LC3-II. The autophagy inhibitor 3-methyladenine (3-MA) markedly inhibited apoptosis, and the apoptosis inhibitor ZVAD-fmk also decreased ISO-induced autophagy. In addition, the PI3K/Akt inhibitor LY294002 enhanced Beclin-1, LC3-II, and poly(ADP-ribose) polymerase (PARP) cleavage levels. Also, the reactive oxygen species (ROS) inhibitor N-acetyl-l-cysteine (NAC), the JNK inhibitor SP600125, and the p38 inhibitor SB203580 efficiently downregulated the levels of these proteins. Moreover, the p53 inhibitor pifithrin-α and the nuclear factor (NF)-κB inhibitor pyrrolidinedithiocarbamic acid (PDTC) clearly suppressed Beclin-1 and LC3-II and increased cytochrome c release, caspase-3 activation, and PARP cleavage. These results demonstrated for the first time that ISO simultaneously induced apoptosis and autophagy by ROS-related p53, PI3K/Akt, JNK, and p38 signaling pathways. Furthermore, ISO-induced apoptosis by activating the Fas receptor-mediated apoptotic pathway and suppressing the p53 and PI3K/Akt-dependent NF-κB signaling pathway, with the subsequent increase in the release of cytochrome c, caspase-3 activation, and PARP cleavage.
BCL-W, BCL-2, BCL-XL MCL-1 INHIBITOR

Isorhamnetin
blocks Hsp70 expression to promote apoptosis
The roots of Codonopis bulleynana Forest ex diels (cbFed), locally known as Tsoong, have been used as a tonic food. Tsoong has wide range of pharmacological effects, including anticancer efficacy. In the present study, the anticancer activity of Tsoong and its potential molecular mechanisms were investigated. Isorhamnetin, a flavonol aglycone, is important compound and metabolite in Tsoong. It can promote apoptosis of colon cancer cells through up-regulating apoptosis-related genes (Apaf1, Casp3 and Casp9) because it blocks Hsp70 genes (Hspa1a, Hspa1b and Hspa8). These were verified by in vitro and in vivo experiments. In vitro, cell counting kit-8 (CCK-8) assays and flow cytometry in HCT116 and SW480 colon cancer cell were used to assess the anti-proliferation and apoptosis-promoting activities of Tsoong. In vivo, the antitumor effect of Tsoong was assessed in colon cancer-bearing nude mice as a xenograft model. These results show that Isorhamnetin is very critical in Tsoong because Tsoong can down-regulate Hsp70 genes and promote apoptosis of colon cancer cells by inhibiting Hsp70 largely due to the efficacy of Isorhamnetin. Our results may ultimately help in the development of diagnostic and therapeutic strategies to control this devastating disease.
Isorhamnetin is an O-methylated flavonol that is predominantly found in the fruits and leaves of various plants, which have been used for traditional herbal remedies. Although several previous studies have reported that this flavonol has diverse health-promoting effects, evidence is still lacking for the underlying molecular mechanism of its anti-cancer efficacy. In this study, we examined the anti-proliferative effect of isorhamnetin on human bladder cancer cells and found that isorhamnetin triggered the gap 2/ mitosis (G2/M) phase cell arrest and apoptosis. Our data showed that isorhamnetin decreased the expression of Wee1 and cyclin B1, but increased the expression of cyclin-dependent kinase (Cdk) inhibitor p21WAF1/CIP1, and increased p21 was bound to Cdk1. In addition, isorhamnetin-induced apoptosis was associated with the increased expression of the Fas/Fas ligand, reduced ratio of B-cell lymphoma 2 (Bcl-2)/Bcl-2 associated X protein (Bax) expression, cytosolic release of cytochrome c, and activation of caspases.Moreover, isorhamnetin inactivated the adenosine 5′-monophosphate-activated protein kinase (AMPK) signaling pathway by diminishing the adenosine triphosphate (ATP) production due to impaired mitochondrial function. Furthermore, isorhamnetin stimulated production of intracellular reactive oxygen species (ROS); however, the interruption of ROS generation using a ROS scavenger led to an escape from isorhamnetin-mediated G2/M arrest and apoptosis. Collectively, this is the first report to show that isorhamnetin inhibited the proliferation of human bladder cancer cells by ROS-dependent arrest of the cell cycle at the G2/M phase and induction of apoptosis. Therefore, our results provide an important basis for the interpretation of the anti-cancer mechanism of isorhamnetin in bladder cancer cells and support the rationale for the need to evaluate more precise molecular mechanisms and in vivo anti-cancer properties.

Isovitexin
induces apoptosis and autophagy
- •Isovitexin inhibits the proliferation and induces apoptosis in human liver cancer.
- •Isovitexin triggers the interplay between apoptosis and autophagy in liver cancer cells.
- •ER stress participates in isovitexin-induced apoptosis and autophagy.
- •Isovitexin displays anti-tumor effect in HepG2 tumor-bearing mice.
Liver cancer is a leading cause of cancer death worldwide, and novel chemotherapeutic drugs to suppress liver cancer are urgently required. Isovitexin(IV), a glycosylflavonoid, is extracted from rice hulls of Oryza sativa, and has various biological activities. However, the anti-tumor effect of IV against liver cancer has not yet been demonstrated in vitro or in vivo. In the present study, we showed that IV significantly suppressed the growth of liver cancer cells. Mechanistic studies indicated that IV induced apoptosis by the mitochondrial apoptotic pathway, as evidenced by the increase of Bax, cleaved Caspase-3, poly (ADP-ribose) polymerase(PARP), and cytoplasm Cyto-c released from mitochondria. In addition, IV resulted in autophagy in liver cancer cells, supported by the enhancement of LC3II, autophagy-related protein (Atg) 3, Atg5 and Beclin1. Suppressing autophagy using bafilomycin A1 (BFA) or siRNA Atg-5 reduced apoptotic cells in IV-treated cells, demonstrating that autophagy induction regulated apoptosis. Moreover, IV was found to cause endoplasmic reticulum (ER) stress in liver cancer cells, along with the promotion of ER stress-related molecules, including inositol-requiring enzyme 1α (IRE1α), X-box-binding protein-1s (XBP-1s), C/EBP homologous protein (CHOP) and glucose-regulated protein (GRP)-78. Of note, inhibition of ER stress by use of its inhibitor, tauroursodeoxycholate (TUDCA), significantly reversed IV-induced apoptosis and autophagy. In vivo, IV treatment showed significant tumor growth inhibition compared to the non-treated group. IV could therefore be a strong candidate for liver cancer prevention.

kaempferol
HDAC INHIBITOR
Kaempferol is a natural polyphenol belonging to the group of flavonoids. Different biological functions like inhibition of oxidative stress in plants or animal cells and apoptosis induction have been directly associated with kaempferol. The underlying mechanisms are only partially understood. Here we report for the first time that kaempferol has a distinct epigenetic activity by inhibition of histone deacetylases (HDACs). In silico docking analysis revealed that it fits into the binding pocket of HDAC2, 4, 7 or 8 and thereby binds to the zinc ion of the catalytic center. Further in vitro profiling of all conserved human HDACs of class I, II and IV showed that kaempferol inhibited all tested HDACs. In clinical oncology, HDAC inhibitors are currently under investigation as new anticancer compounds. Therefore, we studied the effect of kaempferol on human-derived hepatoma cell lines HepG2 and Hep3B as well as on HCT-116 colon cancer cells and found that it induces hyperacetylation of histone complex H3. Furthermore, kaempferol mediated a prominent reduction of cell viability and proliferation rate. Interestingly, toxicity assays revealed signs of relevant cellular toxicity in primary human hepatocytes only starting at 50 μM as well as in an in vivo chicken embryotoxicity assay at 200 μM. In conclusion, the identification of a novel broad inhibitory capacity of the natural compound kaempferol for human-derived HDAC enzymes opens up the perspective for clinical application in both tumor prevention and therapy. Moreover, kaempferol may serve as a novel lead structure for chemical optimization of pharmacokinetics, pharmacology or inhibitory activities.
SASP INHIBITOR
During senescence, cells express molecules called senescence-associated secretory phenotype (SASP), including growth factors, proinflammatory cytokines, chemokines, and proteases. The SASP induces a chronic low-grade inflammation adjacent to cells and tissues, leading to degenerative diseases. The anti- inflammatory activity of flavonoids was investigated on SASP expression in senescent fibroblasts. Effects of flavonoids on SASP expression such as IL-1a, IL-1b, IL-6, IL-8, GM-CSF, CXCL1, MCP-2 and MMP-3 and signaling molecules were examined in bleomycin-induced senescent BJ cells. In vivo activity of apigenin on SASP suppression was identified in the kidney of aged rats. Among the five naturally-occurring flavonoids initially tested, apigenin and kaempferol strongly inhibited the expression of SASP. These flavonoids inhibited NF-kB p65 activity via the IRAK1/IkBa signaling pathway and expression of IkBz. Blocking IkBz expression especially reduced the expression of SASP. A structure-activity relationship study using some synthetic flavones demonstrated that hydroxyl substitutions at C-20,30,40,5 and 7 were important in inhibiting SASP production. Finally, these results were verified by results showing that the oral administration of apigenin significantly reduced elevated levels of SASP and IkBz mRNA in the kidneys of aged rats. This study is the first to show that certain flavonoids are inhibitors of SASP production, partially related to NF-kB p65 and IkBz signaling pathway, and may effectively protect or alleviate chronic low-grade inflammation in degenerative diseases such as cardiovascular diseases and late-stage cancer.
Inhibitory effects of KAEMPFEROL-3-O-sophoroside on HMGB1-mediated proinflammatory responses
High mobility group box 1 (HMGB1) protein is secreted by activated cells of the innate immune system and/or released by injured tissues and necrotic cells; HMGB1 up-regulates proinflammatory cytokines in several inflammatory diseases. Kaempferol-3-O-sophoroside (KP) was isolated from the leaves of cultivated mountain ginseng. KP has antitumor, antioxidative, antiallergic and antidiabetic activities, but the effects of KP on HMGB1-mediated proinflammatory responses have not been studied. In this study, we monitored the effect of KP on the lipopolysaccharide (LPS)-mediated release of HMGB1 and the HMGB1-mediated modulation of proinflammatory responses in human endothelial cells. We found that KP potently inhibited the release of HMGB1 by LPS and inhibited LPS- or HMGB1-mediated barrier permeability and expression of cell adhesion molecules. Further studies revealed that KP inhibited cell surface receptor of HMGB1, toll-like receptor (TLR) 2/4, but not the receptor for advanced glycation end products (RAGE). Collectively, these results suggest that KP possesses anti-inflammatory responses against HMGB1-mediated proinflammatory responses, thereby endorsing its usefulness as a therapy for vascular inflammatory diseases.
► HMGB1 is proinflammatory cytokine as a late mediator of inflammation. ► Kaempferol-3-O-sophoroside (KP) was isolated from mountain ginseng for the first time. ► KP inhibits LPS induced HMGB1 release. ► It also inhibits HMGB1 mediated inflammatory responses.
KAEMPFEROL alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway
•Kaempferol alleviates inflammatory responses induced by LPS in the brain of mice.
•Kaempferol improves blood-brain barrier dysfunction induced by LPS in mice.
•Kaempferol reduces HMGB1 releaseand down-regulates TLR4/MyD88 inflammatory pathway in the brain of mice.
Kaempferol is a natural flavonoid with many biological activities including anti-oxidation and anti-inflammation. Nevertheless, its anti-neuroinflammation role and the relevant mechanism remain unclear. The present study was to investigate effects of kaempferol against LPS-induced neuroinflammation and blood-brain barrier dysfunction as well as the mechanism in mice. BALB/c mice were treated with LPS 5 mg/kg to induce inflammation after pre-treatment with kaempferol 25, 50, or 100 mg/kg for 7 days. The results showed that kaempferol reduced the production of various pro-inflammatory factors and inflammatory proteins including IL-1β, IL-6, TNF-α, MCP-1, COX-2 and iNOS in brain tissues. In addition, kaempferol also protected BBB integrity and increased BBB related proteins including occludin-1, claudin-1 and CX43 in brain of LPS-induced mice. Furthermore, kaempferol significantly reduced HMGB1 level and suppressed TLR4/MyD88 inflammatory pathway in both transcription level and translation level. These results collectively suggested that kaempferol might be a promising neuroprotective agent for alleviating inflammatory responses and BBB dysfunction by inhibiting HMGB1 releaseand down-regulating TLR4/MyD88 inflammatory pathway.
HIF-1α INHIBITOR
Kaempferol has been shown to inhibit vascular formation in endothelial cells. However, the underlying mechanisms are not fully understood. In the present study, we evaluated whether kaempferol exerts antiangiogenic effects by targeting extracellular signal-regulated kinase (ERK)/p38 mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/Akt/mechanistic target of rapamycin (mTOR) signaling pathways in endothelial cells. Endothelial cells were treated with various concentrations of kaempferol for 24 h. Cell viability was determined by the 3- (4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay; vascular formation was analyzed by tube formation, wound healing, and mouse aortic ring assays. Activation of hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor receptor 2 (VEGFR2), ERK/p38 MAPK, and PI3K/Akt/mTOR was analyzed by Western blotting. Kaempferol significantly inhibited cell migration and tube formation in endothelial cells, and suppressed microvessel sprouting in the mouse aortic ring assay. Moreover, kaempferol suppressed the activation of HIF-1α, VEGFR2, and other markers of ERK/p38 MAPK and PI3K/Akt/mTOR signaling pathways in endothelial cells. These results suggest that kaempferol inhibits angiogenesis by suppressing HIF-1αand VEGFR2 activation via ERK/p38 MAPK and PI3K/Akt/mTOR signaling in endothelial cells.
Bcl-2 / BCL-w INHIBITOR
We found that the downregulation of PI3-K, AKT, LC3A/B, MMP9, Bcl-2, and BCL-Wand upregulation of PTEN, p53, p21, and caspase 3 in Kaempferol treated HCC cells. Subsequently, we observed that Kaempferol effectively inhibits cellular proliferation, migration and enhances apoptosis, cell cycle arrest, autophagy, and 5-Fluorouracil mediated chemosensitivity in HCC cells.
INHIBITS EGFR / p38 signaling
Kaempferol has been shown to inhibit cell growth, induce apoptosis and cell cycle arrest in several tumors, but not in renal cell carcinoma (RCC). In the present study, we investigated the effects of kaempferol and the underlying mechanism(s) on the cell growth of RCC cells. MTT assay and colony formation assay were used to study cell growth, and flow cytometry was used to study apoptosis and cell cycles in different RCC cells treated with various doses of kaempferol. A significant inhibition on cell growth, induction of apoptosis and cell cycle arrest were observed in 786-O and 769-P cells after kaempferol treatment compared with the control group. Moreover, the results clearly showed that kaempferol causes a strong inhibition of the activation of the EGFR/p38 signaling pathways, upregulation of p21 expression and downregulation of cyclin B1 expression in human RCC cells, together with activation of PARP cleavages, induction of apoptotic death and inhibition of cell growth. Collectively, our results suggest that kaempferol may serve as a candidate for chemopreventive or chemotherapeutic agents for RCC.
DOWNREGULATES Angiopoietin-like 2 (angptl2)
In murine mastitis, kaempferol (10 or 30 mg/kg) treatment prevented mastitis development, decreased myeloperoxidase (MPO) production, interleukin (IL)-6 level, tumour necrosis factor-α (TNF-α) concentration, and ANGPTL2 expression. In MMEC, kaempferol (1, 3 or 10 μM) reduced MPO production, TNF-α concentration, IL-6 level, and ANGPTL2 expression.
The results in present study show that kaempferol modulates the expression of ANGPTL2to lessen the mastitis in mice.
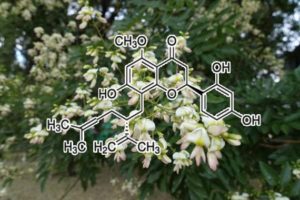
Kurarinone
This study was designed to investigate the effects of the prenylated flavonoid kurarinone on TNF-related apoptosis inducing ligand (TRAIL)-induced apoptosis and its underlying mechanism. A low dose of kurarinone had no significant effect on apoptosis, but this compound markedly promoted tumor cell death through elevation of Bid cleavage, cytochrome c release and caspase activation in HeLa cells treated with TRAIL. Caspase inhibitors inhibited kurarinone-mediated cell death, which indicates that the cytotoxic effect of this compound is mediated by caspase-dependent apoptosis. The cytotoxic effect of kurarinone was not associated with expression levels of Bcl-2 and IAP family proteins, such as Bcl-2, Bcl-xL, Bid, Bad, Bax, XIAP, cIAP-1 and cIAP-2. In addition, this compound did not regulate the death-inducing receptors DR4 and DR5. On the other hand, kurarinone significantly inhibited TRAIL-induced IKK activation, IκB degradation and nuclear translocation of NF-κB, as well as effectively suppressed cellular FLICE-inhibitory protein long form (cFLIPL) expression. The synergistic effects of kurarinone on TRAIL-induced apoptosis were mimicked when kurarinone was replaced by the NF-κB inhibitor withaferin A or following siRNA-mediated knockdown of cFLIPL. Moreover, cFLIP overexpression effectively antagonized kurarinone-mediated TRAIL sensitization. These data suggest that kurarinone sensitizes TRAIL-induced tumor cell apoptosis via suppression of NF-κB-dependent cFLIP expression, indicating that this compound can be used as an anti-tumor agent in combination with TRAIL.

licoricidin
HIF-1A / NFKB INHIBITOR
HIF-1α plays a crucial role in cancer progression by activating the transcription of a broad range of genes involved in the pathophysiology of cancer, including genes implicated in tumor growth, metastasis, angiogenesis, invasion, and metastasis [28,29]. In the present study, the expression of HIF-1α and its downstream targets VEGF-A, COX-2, VEGF-R2, and iNOS [24] were reduced in tumor tissue treated with HEGU or licoricidin (Figures 1C and 4A). In addition, HEGU and licoricidin reduced the expression of phosphorylated p65NFκB. Active NFκB upregulates the expression of genes such as VEGF-C, iNOS, MMP-9, ICAM-1 and VCAM-1that regulate tumor angiogenesis and promote tumor metastasis [30,31]. These results indicate that HEGU and its metastatically active compound licoricidin reduced NFκB and HIF-1α activity, thereby inhibiting the expression of genes involved in the stimulation of tumor angiogenesis.
Modulations of the Bcl-2/Bax Family Were Involved in the Chemopreventive Effects of Licorice Root (Glycyrrhiza uralensis Fisch) in MCF-7 Human Breast Cancer Cells
Recently, cancer chemoprevention with strategies using foods and medicinal herbs has been regarded as one of the most visible fields for cancer control. Genistein in soy, American ginseng, and resveratrol are well-known to have antiproliferative properties in human breast cancer. Licorice root is a botanical, a shrub native to southern Europe and Asia, which primarily has desirable qualities in sweetening and herbal medicine. In this study, licorice (Glycyrrhiza uralensis Fisch) root also inhibits cell proliferation in human breast cancer cell. The cell proliferation study demonstrated that licorice root reduced the proliferation of MCF-7 cells in a dose- and time-dependent manner. The extracts were fractionated in CHCl3, EtOAc, C6H14, and CH3OH−H2O (70:30), and these extracts of licorice root (50 μg/mL) induced DNA fragmentation demonstrated by Hoechst 33258 staining. Apoptosis also determined the sub-G1 accumulation by flow cytometry analysis. These results were consistent with specific cleavage of PARP and antiapoptotic protein Bcl-2 and up-regulation of proapoptotic protein Bax demonstrated by Western blotting. Our findings suggest that licorice root may have chemopreventive effects against human breast cancer through the modulation of the expression of the Bcl-2/Bax family of apoptotic regulatory factors.
Lipoic acid
decreases Mcl-1, Bcl-xL and up regulates Bim on ovarian carcinoma cells leading to cell death
LA suppressed growth proliferation and induced apoptosis in both ovarian cell lines. Moreover, LA provoked a down regulation of two anti-apoptotic proteins, Mcl-1 and Bcl-xLprotein and a strong induction of the BH3-only protein Bim. Furthermore, LA induced ROS generation which could be involved in the CHOP induction which is known to activate the Bim translation.
Conclusion: Our results reveal novel actions of LA which could explain the anti-tumoral effects of the LA. Therefore, LA seems to be a promising compound for ovarian cancer treatment.

Lupeol
up regulation BAX / downregulation BCL-2
Lup-20(29)-en-3h-ol (Lupeol), a triterpene found in fruits such as olive, mango, strawberry, grapes and figs, in many vegetables, and in several medicinal plants, is used in the treatment of various diseases. It possesses strong anti-inflammatory, anti-arthritic, anti-mutagenic, and anti-malarial activity when examined in in vitro and in vivo systems ( Geetha & Varalakshmi 2001)
Study from our laboratory has shown that lupeol (1–30 μM) induced the cleavage of PARP protein and degradation of acinus protein with a significant increase in the expression of FADD protein and Fas receptor in androgen-sensitive human PCa cells. The small interfering RNA-mediated silencing of the Fasgene and inhibition of caspases-6, -8, and -9 by their specific inhibitors confirmed that Lupeol specifically activates the Fas receptor-mediated apoptotic pathway in androgen-sensitive PCa cells. The treatment of cells with a combination of anti-Fas monoclonal antibody and lupeol resulted in higher cell death compared with the additive effect of the two compounds alone, suggesting a synergistic effect. Treatment with lupeol also resulted in a significant inhibition in growth of tumors with concomitant reduction in PSA secretion in athymic nude mice implanted with CWR22Rν1 cells ( Saleem et al. 2005). It has been shown recently that lupeol (400–600 μM) caused anti-proliferative effect associated with an increase in G2/M phase arrest in PC-3 cells. There was also induction of apoptosis with upregulation of Bax, caspases-3 and -9, and APAF-1 genes, and downregulation of anti-apoptotic Bcl-2 gene in PC-3 cells. The role of caspase-induced apoptosis was confirmed by increase in reactive oxygen species, loss of mitochondrial membrane potential followed by DNA fragmentation ( Prasad et al. 2008a; Table 1).
Suppresses C-FLIP
Overexpression of cellular FLICE-like inhibitory protein (cFLIP) is reported to confer chemoresistance in pancreatic cancer (PaC) cells. This study was designed to investigate the effect of lupeol, a dietary triterpene, on (a) apoptosis of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) therapy-resistant PaC cells overexpressing cFLIP and (b) growth of human pancreatic tumor xenografts in vivo. The effect of lupeol treatment on proliferation and TRAIL/caspase-8/cFLIP machinery in PaC cells was investigated. Next, cFLIP-overexpressing and cFLIP-suppressed cells were tested for sensitivity to recombinant TRAIL therapy in the presence of lupeol. Further, athymic nude mice implanted with AsPC-1 cells were treated with lupeol (40 mg/kg) thrice a week and surrogate biomarkers were evaluated in tumors. Lupeol alone treatment of cells caused (a) decrease in proliferation, (b) induction of caspase-8 and poly(ADP-ribose) polymerase cleavage, and (c) down-regulation of transcriptional activation and expression of cFLIP. Lupeol was observed to increase the TRAIL protein level in cells. Lupeol significantly decreased the viability of AsPC-1 cells both in cFLIP-suppressed cells and in cFLIP-overexpressing cells. Lupeol significantly sensitized chemoresistant PaC cells to undergo apoptosis by recombinant TRAIL. Finally, lupeol significantly reduced the growth of human PaC tumors propagated in athymic nude mice and caused modulation of cFLIP and TRAIL protein levels in tumors. Our findings showed the anticancer efficacy of lupeol with mechanistic rationale against highly chemoresistant human PaC cells. We suggest that lupeol, alone or as an adjuvant to current therapies, could be useful for the management of human PaC.

Luteolin
Luteolin promotes degradation in STAT3 in human hepatoma cells: an implication for the antitumor potential of flavonoids.
In this study, we have investigated the underlying molecular mechanism for the potent proapoptotic effect of luteolin on human hepatoma cells both in vitro and in vivo, focusing on the signal transducer and activator of transcription 3 (STAT3)/Fas signaling. A clear apoptosis was found in the luteolin-treated HLF hepatoma cells in a time- and dosage-dependent manner. In concert with the caspase-8 activation by luteolin, an enhanced expression in functional Fas/CD95 was identified. Consistent with the increased Fas/CD95 expression, a drastic decrease in the Tyr(705) phosphorylation of STAT3, a known negative regulator of Fas/CD95 transcription, was found within 20 minutes in the luteolin-treated cells, leading to down-regulation in the target gene products of STAT3, such as cyclin D1, survivin, Bcl-xL, and vascular endothelial growth factor. Of interest, the rapid down-regulation in STAT3 was consistent with an accelerated ubiquitin-dependent degradation in the Tyr(705)-phosphorylated STAT3, but not the Ser(727)-phosphorylated one, another regulator of STAT3 activity. The expression level of Ser(727)-phosphorylated STAT3 was gradually decreased by the luteolin treatment, followed by a fast and clear down-regulation in the active forms of CDK5, which can phosphorylate STAT3 at Ser(727). An overexpression in STAT3 led to resistance to luteolin, suggesting that STAT3 was a critical target of luteolin. In nude mice with xenografted tumors using HAK-1B hepatoma cells, luteolin significantly inhibited the growth of the tumors in a dosage-dependent manner. These data suggested that luteolin targeted STAT3 through dual pathways-the ubiquitin-dependent degradation in Tyr(705)-phosphorylated STAT3 and the gradual down-regulation in Ser(727)-phosphorylated STAT3 through inactivation of CDK5, thereby triggering apoptosis via up-regulation in Fas/CD95.
INHIBITS HMGB1 & HSP90
•Luteolin inhibits the secretion of HMGB1 and aggravation of inflammatory cascades.
•Luteolin destabilizes the protein levels of c-Jun and Akt (Hsp90 client proteins).
•Luteolin protects against LPS induced lethality in vivo.
Septic diseases represent the prevalent complications in intensive care units. Luteolin, a plant flavonoid, has potent anti-inflammatory properties; however, the molecular mechanism beneath luteolin mediated immune modulation remains unclear. Here in vitro investigations showed that luteolin dose-dependently inhibited LPS-triggered secretion and relocation of high mobility group B-1 (HMGB1) and LPS-induced production of tumor necrosis factor alpha (TNF-α) and nitric oxide (NO) in macrophages. The mechanism analysis demonstrated that luteolin reduced the release of HMGB1 through destabilizing c-Jun and suppressed HMGB1-induced aggravation of inflammatory cascade through reducing Akt protein level. As an inhibitor ofHsp90, luteolin destabilized Hsp90 client protein c-Jun and Akt. In vivo investigations showed that luteolin effectively protected mice from lipopolysaccharide (LPS)-induced lethality. In conclusion, the present study suggested that luteolin may act as a potential therapeutic reagent for treating septic diseases.
One clear potential therapeutic target for AD has been gamma-secretase. Given the obvious implications on gamma-secretase activity, we analysed what effect luteolin treatment had on SweAPP N2a cells using a fluorometric assay for gamma-cleavage. In accordance with expectations, luteolin lowered gamma-secretase cleavage activity in both a concentration and time-dependent fashion. More importantly, these concentration and time-dependent decreases in gamma-secretase cleavage activity correlate with the decreases in total Aβ generation. Taken together, the above data suggest that luteolin exerts its anti-amyloidogenic effects through down-regulation of gamma-secretase activity.
LUTEOLIN treatment significantly inhibited cell senescence and increased telomerase activity
Effect of Luteolin and Apigenin on the Cell Senescence and Telomerase Activity of DPCs at Various Passages. Senescenceassociated b-galactosidase is caused by upregulated lysosomal activities and altered cytosolic pH, which are upregulated with senescence and aging. To elucidate the effect of luteolin and apigenin on replicative senescence state of DPCs, the senescence-associated b-galactosidase activity (SA-b-gal) was evaluated. DPCs from passages 1, 3, 5, and 7 with/without luteolin/apigenin treatment were detected, albeit only the representative results of passages 3 and 7 were presented. The result revealed that DPCs at passage 3 with luteolin/apigenin induction and the control group did not show any obvious blue staining. DPCs at passage 7 without induction showed intense blue color, albeit DPCs at passage 7 with luteolin or apigenin induction revealed weak blue staining, not as intense as the control group at passage 7. Similarly, there is no difference of the telomerase activity of DPCs at passage 3 with/without luteolin or apigenin induction albeit DPCs at passage 7 with luteolin or apigenin induction showed significantly higher telomerase activity than the control group at passage 7 (∗ 𝑃 < 0.05), which agreed with the result of b-galactosidase assay mentioned above. This result implied that luteolin and apigenin treatment significantly inhibited cell senescence and increased telomerase activityof DPCs, especially at late passages. Thus, luteolin and apigenin might be able to maintain DPCs in an undifferentiated and presenescent state.
acetyltransferase p300 inhibitor
Luteolin was found to inhibit p300 acetyltransferase with competitive binding to the acetyl CoA binding site. Luteolin treatment in a xenografted tumor model of head and neck squamous cell carcinoma (HNSCC), led to a dramatic reduction in tumor growth within 4 weeks corresponding to a decrease in histone acetylation. Cells treated with luteolin exhibit cell cycle arrest and decreased cell migration. Luteolin treatment led to an alteration in gene expression and miRNA profile including up-regulation of p53 induced miR-195/215, let7C; potentially translating into a tumor suppressor function. It also led to down-regulation of oncomiRNAs such as miR-135a, thereby reflecting global changes in the microRNA network. Furthermore, a direct correlation between the inhibition of histone acetylation and gene expression was established using chromatin immunoprecipitation on promoters of differentially expressed genes. A network of dysregulated genes and miRNAs was mapped along with the gene ontology categories, and the effects of luteolin were observed to be potentially at multiple levels: at the level of gene expression, miRNA expression and miRNA processing.
MDM2 INHIBITOR
Luteolin binds with highest negative binding energy value and shows highest potency towards MDM2 inhibition.Although, the MDM2 residues interacting with polyphenols were the same for all but, the change in groove structure significantly affected the position of polyphenol in the groove and also its binding energy. The hydrophobic interactions were solely responsible for stable com- plex formation as revealed by the vdW energy and ligplot analysis. Finally, on the basis of data obtained during the study, it can be concluded that these polyphenols have the potential to be used as lead molecules for the inhibition of MDM2 with Luteolin being the top candidate. This approach can be used to screen out a huge number of natural compounds for their potency in anti-cancer treatment.
Lycopene suppresses proinflammatory response in lipopolysaccharide-stimulated macrophages by inhibiting ROS-induced trafficking of TLR4to lipid raft-like domains
We recently showed that lycopene inhibited lipopolysaccharide (LPS)-induced productions of nitric oxide (NO) and interleukin-6 (IL-6) in murine RAW264.7 macrophages by mechanisms related to inhibition of ERK and nuclear factor-κB. Since the assembly of Toll-like receptor 4 (TLR4) in lipid rafts is a key element in LPS induced signaling, we investigated whether this process would be influenced by lycopene. We found that pretreatment of RAW264.7 cells with lycopene inhibited LPS-induced recruitment of TLR4into fractions — enriched with lipid raft marker. By the methods of immunoprecipitation and immunoblotting, we also found that lycopene inhibited the subsequent formation of the complex of TLR4 with its adaptors including myeloid differentiation primary-response protein 88 and TIR domain–containing adaptor-inducing IFN-β. We also found that the lycopene induced inhibition was associated with reduced formation of reactive oxygen species (ROS), which was an upstream mechanism for the effects of lycopene, because treating the cells with the antioxidant N-acetyl-l-cysteine and NADPH oxidase inhibitor diphenyleneiodonium chloride significantly inhibited LPS-induced recruitment of TLR4 into lipid raft-like domains as well as the production of proinflammatory molecule NO and IL-6. Thus, our findings suggest that lycopene may prevent LPS-induced TLR4 assembly into lipid rafts through reducing intracellular ROS level.
NFKB INHIBITOR
The amelioration of Aβ1–42-induced spatial learning and memory impairment by lycopene could be linked, at least in part, to the inhibition of NF-κB activity and the down-regulation of expression of neuroinflammatory cytokines, suggesting that lycopene may be a potential candidate for AD treatment.
Combination chemoprevention by diet-derived agents that induce apoptosis is a promising strategy to control gastric cancer, the second most common malignancy worldwide. The present study was undertaken to investigate the apoptosis-inducing potential of a combination of S-allylcysteine (SAC), an organosulphur constituent of garlic and lycopene, a tomato carotenoid during N-methyl-N′-nitro-N–nitroso-guanidine (MNNG) and saturated sodium chloride (S-NaCl)-induced gastric carcinogenesis in Wistar rats using the apoptosis-associated proteins Bcl-2, Bax, Bim, caspase 8 and caspase 3 as markers. Animals administered MNNG followed by S-NaCl developed squamous cell carcinomas of the stomach associated with increased Bcl-2 expression and decreased expression of Bax, Bim, caspase 8 and caspase 3. Although SAC and lycopene alone significantly suppressed the development of gastric cancer, administration of SAC and lycopene in combination was more effective in inhibiting MNNG-induced stomach tumours and modulating the expression of apoptosis-associated proteins. Our results suggest that induction of apoptosis by SAC and lycopenecombination represents one of the possible mechanisms that could account for their synergistic chemopreventive activity against gastric cancer.
p21/Cip1↑, p27/Kip1↑, Hypoxia⊥, HIF1α↑, VEGF↑, AMPK↑, Bcl2↓, Bax↑, p53↑, Bax/Bcl-2↑, caspase-3↑, cyclin B1↓, cyclin A↓, CDK-4↓, Cdc2↓, Cip↑, caspase-8↑, PARP↑, NF-κB↓, HER2↓, PI3K/Akt↓, Bad↑, Bcl-X(S)↑, Bcl-X(L)↓, MMP-2↓, MMP-9↓, caspase-3, -9↑, ERK↑, Raf-1↑, Ca(2+)↑, Cyto-c↑, bcl-2↓, LTC4⊥, LTB4⊥, IgE⊥, cPLA2⊥, 5-LO⊥, MMP-9⊥, Ca(2+)↑, caspase-7↑, ADP- ribose↓, phosphatase↑
Although hepatitis B and/or hepatitis C virus were recognized as major risk factor for the development of hepatocellular carcinoma (HCC), certain occupational, environmental, and lifestyle factors also play key roles in HCC tumorigenesis. Moreover, in molecular signaling route, extracellular signal‐regulated kinase (ERK)/nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) signaling was found to be overexpressed and linked to poor prognosis in HCC. Thus, to identify possible nature compound that can suppress ERK/NF‐κB may be benefit to HCC patient. Magnolol, a natural compound derived from herbal plant Magnolia officinalis, has been recognized as a liver protection and antitumor reagent. However, whether magnolol‐inhibited HCC progression correlates with disruption of ERK/NF‐κB signaling is remained unclear. In this studies, we performed SK‐Hep1/luc2 HCC bearing animal model to investigate the anticancer efficacy and mechanism of magnolol on tumor progression. Tumor size and tumor growth rate were dramatically suppressed after treatment of magnolol. In addition, expression of phospho‐ERK (p‐ERK), NF‐κB p65 (Ser536), and tumor progression‐associated proteins, such as matrix metallopeptidase 9 (MMP‐9), vascular endothelial growth factor (VEGF), X‐linked inhibitor of apoptosis protein (XIAP), and CyclinD1 were all significantly decreased by magnolol. Most important, major extrinsic and intrinsic apoptosis signaling factors, including active caspase‐8 and caspase‐9 were both enhanced by magnolol. This study indicated that apoptosis induction through extrinsic/intrinsic pathways and blockage of ERK/NF‐κB activation were associated with magnolol‐inhibited tumor progression in HCC in vivo.

CDK2 INHIBITOR
This study was carried out to investigate the impact of polyphenol-rich extract from mulberry leaf on the proliferation of vascular smooth muscle cell (VSMC) and verify its mechanism in vitro. VSMC proliferation is an important pathophysiological process in the development of atherosclerosis, which is the major cause of coronary artery disease (CAD). Polyphenol-rich foods, such as mulberry leaf, have been reported to reduce the risk of CAD. The effect of mulberry leaf extract (MLE) on cell growth was measured by a growth curve assay, on distribution of cells in the cell cycle by flow cytometry, and on cyclin-dependent kinase (CDK) activity and cell-cycle regulatory proteins by Western blot, immunoblotting, and immunoprecipitation analyses. The results showed that MLE induced phosphorylation of p53, promoted expression of p21 and p27, decreased CDK2/4 activity,inhibited phosphorylation of Rb, and thereby blocked the G1 to S transition in the cell cycle.
down-regulation of NFκB and its target genes Bcl-2 & Bcl-xl
In this study, the antioxidative fraction of white mulberry (Morus alba) was found to have an apotogenic effecton Ehrlich’s ascites carcinoma cell-induced mice (EAC mice) that correlate with upregulated p53 and downregulated NFκB signaling. The antioxidant activities and polyphenolic contents of various mulberry fractions were evaluated by spectrophotometry and the ethyl acetate fraction (EAF) was selected for further analysis. Strikingly, the EAF caused 70.20% tumor growth inhibition with S-phase cell cycle arrest, normalized blood parameters including red/white blood cell counts and suppressed the tumor weight of EAC mice compared with untreated controls. Fluorescence microscopy analysis of EAF-treated EAC cells revealed DNA fragmentation, cell shrinkage, and plasma membrane blebbing. These characteristic morphological features of apoptosis influenced us to further investigate pro- and anti-apoptotic signals in EAF-treated EAC mice. Interestingly, apoptosis correlated with the upregulation of p53 and its target genes PARP-1 and Bax, and also with the down-regulation of NFκB and its target genes Bcl-2 and Bcl-xL. Our results suggest that the tumor- suppressive effect of the antioxidative fraction of white mulberry is likely due to apoptosis mediated by p53 and NFκB signaling.
AKT / MTOR INHIBITOR
Hepatocellular carcinoma (HCC) is one of the most common malignancies in Taiwan. Many risks factors induce liver chronic inflammation, fibrosis, cirrhosis, and hepatocellular carcinoma. Mulberry fruits containing polyphenols to remove free radicals and mitigate inflammation has been reported to not only against gastric cancer, melanoma and leukemia but also prevent liver injury induced by alcohol or CCl4 in previous researches. The aim of this study is to examine whether Mulberry could inhibit hepatocarcinogenesis. In animal experiment, diethylnitrosamine (DEN) was used to induce hepatic tumorgenesis. After injecting DEN, the rats treated with mulberry water extracts (MWE) had less and smaller tumor than others without MWE. Moreover, MWE reduced the serum ALT and AST, HCC marker, cleavage caspases, Ser-15-p53 and Ser46-p53 induced by DEN. Further, we observed that mulberry polyphenol extracts (MPE) inhibited the cell growth of HepG2 cell and Hep3B cell. By using flow cytometry and western blotting methods, MPE induced HepG2 cell apoptosis by increase subG1 cells and the elevated expression of caspase-3/8/9. Instead of apoptosis, MPE caused Hep3B cells autophagy by inhibiting Akt and mTOR phosphorylation. Comprehensively, mulberry extracts has a potential to be a health supplement to prevent hepatocarcinogenesis in the future.
Natural killer (NK) cells are capable of identifying and killing tumor cells as well as virus infected cells without pre-sensitization. NK cells express activating and inhibitory receptors, and can distinguish between normal and tumor cells. The present study was designed to demonstrate the importance of the expression level of NKG2D ligands on the Burkitt’s lymphoma cell line, Raji, in enhancing NK cell cytolytic activity. Various flavonoids were used as stimulants to enhance the expression of NKG2D ligands. NK cell lysis activity against Raji was not changed by pre-treatment of Raji with luteolin, kaempferol, taxifolin and hesperetin. However, treatment of Raji with naringenin showed increased sensitivity to NK cell lysis than untreated control cells. The activity of naringenin was due to enhanced NKG2D ligand expression.These results provide evidence that narigenin’s antitumor activity may be due to targeting of NKG2D ligand expression and suggests a possible immunotherapeutic role for cancer treatment.
myricetin
Induces Apoptosis via Suppressing PI3K/Akt/mTOR
Apoptosis is a highly regulated process; the tumor suppressor protein, p53, induces as well as inhibits apoptosis.39 It regulates DNA repair and cell cycle progression.40 The induction of apoptosis by myricetin was confirmed by an increase in the percentage of annexin V-positive cells, the levels of p53 and Bax, and the cleavage of procaspase-3 to caspase-3. Oxidative stress occurs due to an imbalance between pro-oxidant and antioxidant cellular factors, and causes cell damage.41 ROS comprise superoxide and hydroxyl radicals, hydrogen peroxide, and singlet oxygen; they are byproducts of mitochondrial respiration and contribute to oxidative stress. Increased intracellular levels of ROS lead to the activation of apoptotic pathways.42 Upon analysis using an oxidant-sensitive fluorescent dye (H2DCFDA), we found that myricetin significantly enhanced the production of intracellular ROS in HUVECs, indicating that it induces apoptosis by increasing intracellular ROS levels. Mitochondria play a crucial role in respiratory metabolism and cell cycle progression; they regulate extrinsic and intrinsic apoptotic pathways, and affect cell proliferation, differentiation, and survival. They are the major cell organelles responsible for intracellular ROS production. Increased ROS levels reduce the mitochondrial membrane potential, thereby activating apoptotic pathways; therefore, mitochondrial dysfunction leads to apoptosis.43 The PI3K/Akt/mTOR pathway and its downstream targets are activated during angiogenesis.44 Akt is a serine/threonine kinase that regulates a variety of cellular functions, including cell growth, proliferation, migration, protein synthesis, transcription, survival, and angiogenesis.45 mTOR kinase, the central regulator of cell metabolism, growth, proliferation, and survival, is also activated during tumor initiation, progression, and angiogenesis.9,45
In conclusion, the findings of this study suggest that myricetin inhibits angiogenesis in endothelial cells by inducing ROS-mediated apoptosis and inhibiting PI3K/Akt/mTOR signaling. Therefore, we propose that myricetin, as an antiangiogenic agent, is a promising anticancer drug candidate.
INHIBITS BCL-2 & BCL-XL
The expression of Puma, Bcl-2, Bcl-xl, Bax, Bad and caspase-9 proteins were detected by western blotting. As shown in Fig. 4A, the expression of Bax and Bad proteins was increased, while the Bcl-2 and Bcl-xl protein levels were decreased when treated with myricetin
down-regulatES C-FLIP and bcl-2
The flavonoid Myricetin has been reported to exhibit therapeutic activity in cancer. In this study glioblastoma cells and human astrocytes were treated with Myricetin, tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) or the combination of both. Treatment with subtoxic doses of Myricetin in combination with TRAIL induces rapid apoptosis in glioma cells. Notably, human astrocytes were not affected by the combined treatment consisting of Myricetin and TRAIL. Combined treatment with Myricetin and TRAIL augmented the activation of initiator caspases-8/-9 and effector caspases-3/-7. Furthermore, Myricetin down regulated the expression of the long and short isoform of c-FLIP and bcl-2and over-expression of the short isoform of c-FLIP (S) and bcl-2 attenuated apoptosis induced by the combination of Myricetin and TRAIL. Furthermore, Myricetin did not modulate the mRNA levels of c-FLIP, suggesting that Myricetin modulates the expression of c-FLIP in a posttranscriptional manner. In summary, the short isoform of c-FLIP and bcl-2 are key regulators in TRAIL-Myricetin mediated cell death in malignant glioma.
Sulforaphane and myricetin act synergistically to induce apoptosis in 3T3‑L1 adipocytes
The aim of the present study was to investigate whether sulforaphane (SFN) and myricetin (Myr) synergistically induce apoptosis in adipocytes. The viability of mature 3T3‑L1 adipocytes treated with 40 µM SFN and/or 100 µM Myr was assessed using an MTT assay. Apoptosis was assessed by Hoechst 33258 nuclear staining, and by detection of single‑stranded DNA using an enzyme‑linked immunosorbent assay. Compared with the effects of each compound alone, the combination of SFN and Myr synergistically reduced cell viability, induced apoptosis, increased pro‑apoptotic Bcl‑2 associated X protein expression, decreased anti‑apoptotic B‑cell lymphoma‑2 expression, enhanced Bcl‑2‑associated death promoter (Bad) translocation from the cytoplasm to the mitochondria, and reduced Bad phosphorylation at Ser112. These effects were accompanied by increased cleavage of caspase 3 and poly‑ADP‑ribose‑polymerase. In addition, combined SFN and Myr treatment significantly decreased the protein expression levels of phosphorylated AKT serine/threonine kinase 1 (Akt) at Ser473, as well as the phosphorylation of the downstream protein ribosomal protein, S6 kinase β‑1. Therefore, SFN plus Myr was a more potent inducer of apoptosis in 3T3‑L1 adipocytes than either compound alone. The results of the present study suggest that the mechanism of SNF/Myr‑induced apoptosis involved activation of the Akt‑mediated mitochondrial apoptotic pathway. This may aid treatment of animal models of obesity and preclinical testing.

naringenin
Inhibitory effect of naringenin on LPS-induced skin senescence by SIRT1 regulation in HDFs
This study confirmed that naringenin inhibits cellular senescence and regenerates human dermal fibroblasts damaged by lipopolysaccharide, and suggests that naringenin will be a cosmetic ingredient that has cell regenerative effects and anti-aging effects. In conclusion, this in vitro research confirmed that naringenin not only increases SIRT1 activity but, by blocking the NF-κB pathway, also inhibits the gene expression of inflammatory factors (which are major components of SASPs) and substances that produce ROS. Naringenin can be used as an effective cosmetic ingredient to prevent age-induced skin elasticity reduction and wrinkled skin, troublesome skin diseases, and premature skin aging caused by harmful environmental pollutants, PM, etc.
ACTIVATES NRF2
Oxidative stress and mitochondrial dysfunction are considered to be major contributing factors in the development and progression of many neurodegenerative diseases. Naringenin (NAR) is an abundant flavanone in the Citrus genus and has been found to exert antioxidant, anticarcinogenic and antimutagenic effects. However, the potential underlying mechanism of its antioxidant effects remains unclear. In the present study, the authors investigated the antioxidant effect of NAR on neurons in vitro. Neurons isolated from the brains of Sprague-Dawley rats were randomly divided into a control group, model group, NAR-L group, NAR-M group and NAR-H group. The model group received hypoxia and re-oxygenation treatment, and the NAR-L, NAR-M and NAR-H groups received 20, 40 and 80 µM NAR, respectively. The levels of reactive oxygen species (ROS) in each group were detected by chloromethyl-2′,7’dichlorodihydro fluorescein diacetate staining, and differences in mitochondrial dysfunction were analyzed through measurement of mitochondrial membrane potential (∆ψm), adenine nucleotide translocase transport activity and adenine nucleotide levels. MTT and flow cytometry assays were also used to analyze cell proliferation and apoptosis, and the effects of NAR on the nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE) signaling pathway were investigated using small interfering RNA methods. The authors detected an increased accumulation of ROS in the model group, and high-dose NAR could significantly reduce the levels of ROS. Furthermore, NAR could improve mitochondrial dysfunction, as indicated by increased levels of high-energy phosphates, enhanced mitochondrial ANT transport activity and increased mitochondrial membrane potential. Moreover, NAR increased cell viability and decreased the rate of cell apoptosis. NAR also increased the expression of Nrf2 and its downstream target genes. These findings demonstrated that NAR could reduce oxidative stress and improve mitochondrial dysfunction via activation of the Nrf2/ARE signaling pathway in neurons.
downregulates Bcl 2, Mcl 1 and Btk
The results revealed that naringenin downregulated the newlineexpression of Bcl 2, Mcl 1 and Btk proteins, while upregulating cleaved caspase 3 in CLL patient samples, newlinethereby controlling BCR signaling pathway, and also triggers apoptosis by intrinsic pathway.
ENHANCES NKG2D LIGAND EXPRESSION
Induction of apoptosis is one of the most active strategies in cancer chemoprevention and the ability of medicinal plants in this regard has attracted major research interest. The present study was designed to investigate the apoptosis inducing capacity of an ethanolic neem leaf extract (ENLE) during 7,12-dimethylbenz[a]anthracene (DMBA)-induced hamster buccal pouch carcinogenesis using the apoptosis-associated proteins Bcl-2, Bim, caspase 8 and caspase 3 as markers. Topical application of DMBA to the hamster cheek pouch for 14 weeks resulted in well developed squamous cell carcinomas associated with increased expression of Bcl-2 and decreased expression of Bim, caspase 8 and caspase 3. Administration of ENLE inhibited DMBA-induced hamster buccal pouch (HBP) carcinogenesis, as revealed by the absence of neoplasms, with induction of Bim and caspases 8 and 3 and inhibition of Bcl-2 expression. Our results suggest that the chemopreventive effects of ENLE may be mediated by induction of apoptosis.
Key Words: Apoptosis – Bcl-2 – Bim – caspases – hamster buccal pouch carcinogenesis – neem extract Asian Pacific J Cancer Prev, 6, 515-520
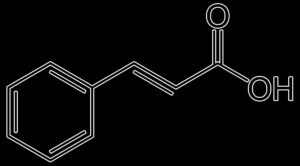
Oleacein
HDAC INHIBITOR
Olive oil contains different biologically active polyphenols, among which oleacein, the most abundant secoiridoid, has recently emerged for its beneficial properties in various disease contexts. By using in vitro models of human multiple myeloma (MM), we here investigated the anti-tumor potential of oleacein and the underlying bio-molecular sequelae. Within a low micromolar range, oleacein reduced the viability of MM primary samples and cell lines even in the presence of bone marrow stromal cells (BMSCs), while sparing healthy peripheral blood mononuclear cells. We also demonstrated that oleacein inhibited MM cell clonogenicity, prompted cell cycle blockade and triggered apoptosis. We evaluated the epigenetic impact of oleacein on MM cells, and observed dose-dependent accumulation of both acetylated histones and α-tubulin, along with down-regulation of several class I/II histone deacetylases (HDACs) both at the mRNA and protein level, providing evidence of the HDAC inhibitory activity of this compound; conversely, no effect on global DNA methylation was found. Mechanistically, HDACs inhibition by oleacein was associated with down-regulation of Sp1, the major transactivator of HDACs promoter, via Caspase 8 activation. Of potential translational significance, oleacein synergistically enhanced the in vitro anti-MM activity of the proteasome inhibitor carfilzomib. Altogether, these results indicate that oleacein is endowed with HDAC inhibitory properties, which associate with significant anti-MM activity both as single agent or in combination with carfilzomib. These findings may pave the way to novel potential anti-MM epi-therapeutic approaches based on natural agents.
Our results indicate that oleacein, the most abundant EVOO secoiridoid, elicits significant anti-tumor activity by promoting cell cycle arrest and apoptosis, either as a single agent or in combination with the proteasome inhibitor carfilzomib. Moreover, our data highlight an epigenetic impact of oleacein in MM, as demonstrated by the impairment of the MM acetylome, likely via Sp1-dependent transcriptional inhibition of HDACs. Altogether, these findings provide the molecular rationale for potential epi-therapeutic anti-MM strategies based on natural agents.
Oleanolic acid
suppression of NF-κB induced Bcl-2 Cyclin D / upregulateS p21 and p27
The objective of this study was to assess the effect of oleanolic acid, a triterpene on inducing apoptosis in B16F-10 melanoma cells. Treatment of B16F-10 cells with nontoxic concentration of oleanolic acid showed the presence of apoptotic bodies and induced DNA fragmentation in a dose depended manner. Cell cycle analysis and TUNEL assay were also confirmed the observation. The apoptotic genes p53, Bax, Caspase-9 and caspase-3 were found upregulated in oleanolic acid treated cells, whereas the anti-apoptotic gene Bcl-2 was downregulated. Oleanolic acid treatment also showed a downregulation of Cyclin D1 expression and upregulated p21 and p27gene expression in B16F-10 melanoma cells. Oleanolic acid inhibited the activation and nuclear translocation of transcription factors such as NF-κB, c-fos, ATF-2 and CREB-1, and also down regulated the production and expression of TNF-α, IL-1β, IL-6 and GM-CS. These results suggest that oleanolic acid induces apoptosis through activating p53 induced caspase-mediated pro-apoptotic pathway and suppression of NF-κB induced Bcl-2mediated anti-apoptotic pathway.
Caveolin-1 key role in the oleanolic acid-induced apoptosis
Our previous study found that caveolin-1 (CAV-1) protein expression is upregulated during oleanolic acid (OA)-induced inhibition of proliferation and promotion of apoptosis in HL-60 cells. CAV-1 is the main structural protein component of caveolae, playing important roles in tumorigenesis and tumor development. It has been shown that cav-1 expression is lower in leukemia cancer cell lines SUP-B15, HL-60, THP-1 and K562 and in chronic lymphocytic leukemia primary (CLP) cells when compared with normal white blood cells, with the lowest cav-1 expression level found in HL-60 cells. To study the effects of cav-1 in HL-60 cells and the effects of cav-1 overexpression on OA drug efficacy, cav-1 was overexpressed in HL-60 cells using lentiviral-mediated transfection combined with OA treatment. The results showed that cav-1 overexpression inhibited HL-60 cell proliferation, promoted apoptosis, arrested the cell cycle in the G1 phase and inhibited activation of the PI3K/AKT/mTOR signaling pathway. Overexpression of CAV-1 also increased HL-60 cell sensitivity to OA. To further verify whether OA affects HL-60 cells via the activation of downstream signaling pathways by CAV-1, cav-1 gene expression was silenced using RNAi, and the cells were treated with OA to examine its efficacy. The results showed that after cav-1 silencing, OA had little effect on cell activity, apoptosis, the cell cycle and phosphorylation of HL-60 cells. This study is the first to show that CAV-1 plays a crucial role in the effects of OA on HL-60 cells.
OA can block cell cycle progression through ERK-P53, promote apoptosis through the mitochondrial pathway, inhibit the mTOR signaling pathway (7) or downregulate Bcl-2 to upregulate Bax and Bad to achieve antitumor effects (8); VEGF is also involved in the OA inhibitory effects on proliferation, invasion and tube formation by endothelial cells (10).
Oleuropein
(a glycosylated seco-iridoid, a type of phenolic bitter compound found in green olive skin, flesh and seeds, leaves, and argan oil.The term oleuropein is derived from the botanical name of the olive tree, Olea europaea.)
oleuropein shows a potent senolytic and anti-inflammatory effect by reducing IL-1β, IL-6 and COX-2 gene expression and NF-kB activation.
Osteoarthritis (OA) is the most prevalent disorder of articulating joints and a leading cause of disability in humans, affecting half of the world’s population aged 65 years or older. Articular cartilage and synovial tissue from OA patients show an overactivity of the membrane channel protein connexin43 (Cx43) and accumulation of senescent cells associated with disrupted tissue regeneration. We have recently demonstrated the use of the Cx43 as an appropriate therapeutic target to halt OA progression by decreasing the accumulation of senescent cells and by triggering redifferentiation of osteoarthritic chondrocytes (OACs) into a more differentiated state, restoring the fully mature phenotype and cartilage regeneration. In this study we have found that small molecular polyphenols derived by olive extracts target Cx43 and senescence in OACs, synovial and bone cells from patients and in human mesenchymal stem cells (hMSCs). Our results indicate that these small molecules including oleuropein regulate the promoter activity of Cx43 gene. The downregulation of Cx43 expression by oleuropein reduce gap junction intercellular communication, cellular senescence in chondrocytes and enhance the propensity of hMSCs to differentiate into chondrocytes and bone cells, reducing adipogenesis. In concordance with these results, these small molecules reduce Cx43 and decrease Twist-1 activity leading to redifferentiation of OACs, which restores the synthesis of cartilage ECM components (Col2A1 and proteoglycans) and reduces inflammatory and catabolic factors IL-1β, IL-6, COX-2 and MMP-3 and cellular senescence orchestrated by p53/p21 together with the synthesis of SASP via NF-kB. Altogether, our results demonstrate the use of the olive-derived polyphenols such as oleuropein as potentially effective therapeutic agents to enhance the efficacy of hMSC therapy and to induce a pro-regenerative environment in OA patients by restoring cellular phenotype and clearing out senescent cells in joint tissues in order to stop or prevent the progression of the disease.
Consistent with previous studies, oleuropein shows a potent senolytic and anti-inflammatory effect by reducing IL-1β, IL-6 and COX-2 gene expression and NF-kB activation119. Our study is the first to demonstrate the effect of this polyphenol on Cx43 activity and senescence. Our results show that oleuropein downregulates Cx43 in OACs, enhancing chondrocyte redifferentiation, together with a decrease in cellular senescence and SASP activated by NF-kB (Fig. 5). Besides, modulation of Cx43 by oleuropein shifts the hMSCs differentiation capacity towards osteogenic and chondrogenic lineages, while decreasing adipogenic differentiation. These effects may be useful in cell therapy approaches that aim to promote cartilage and bone regeneration because the osteogenesis/adipogenesis switch has been associated with different bone disorders characterized by reduced bone formation and increased bone marrow fat accumulation99,100. Altogether, these findings indicate that oleuropein controls Cx43 gene expression and acts as a potent senolytic drug that may serve as a potential agent to improve both the effectiveness of stem cell therapy and cartilage and joint regeneration in patients to stop OA progression, by restoring tissue regeneration.
Oleuropein, a phenolic compound extracted from olive oil and olive tree leaves, has been reported to exert a variety of beneficial effects (Santiago-Mora et al., 2011; Casamenti and Stefani, 2017), including anti-inflammatory, antioxidant, anticancer, and cardio- and neuroprotective actions (Sun et al., 2017a,b). An in vitro study found that its anti- aging effect is mainly mediated by the induction of autophagy in relation to increased AMPK phosphorylation and mTOR inhibition (Rigacci et al., 2015). Treatment of cultured endothelial progenitor cells with oleuropein or oleacein, a derivative of oleuropein, after senescence induction with angiotensin II was associated with a significant reduction in SA β-gal activity and ROS concentration and with increased cell replication and telomerase activity (Santiago-Mora et al., 2011). Treatment of pre-senescent human lung (MRC5) and neonatal human dermal (NHDF) fibroblasts with oleuropein aglycone (OLE) for 4–6 weeks reduced β-gal-positive cells, p16 protein expression, IL-6 and metalloprotease secretion, and COX type 2 (COX-2) and α-smooth actin levels (Menicacci et al., 2017).
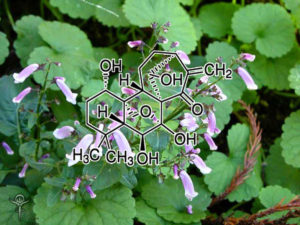
Caspase-8↓, NF-κB (p65)↓, IKKα↓, IKKβ↓, phospho-mTOR↓, Fas↑, PPARγ↑, MMP-2/MMP-9↓, β1/FAK⊥, caspase-3↑, LYN⊥, ABL⊥, Akt/mTOR↓, Raf/MEK/ERK↓ and STAT5↓, AML1-ETO↓, c-Kit(+)⊥, c-Met-NF-κB-COX-2↑, c-Met-Bcl-2–caspase-3, Bcl-2/Bax ratio↑, AVOs↓, LC3-I⊥, LC3-II⊥, P21↑, FAS⊥, SREBP1⊥, AP-1↓, NF-κB↓, P38↓, p21↑, p27↑, p16↑, c-myc p38↑, p53↑, (MAPK)-p38, cyclin B1 and p-cdc2 (T161)↓, p53↑, Akt↓, ROS↓, SIRT1↓, NF-κB↑, caspase-1↑, IL1β↑, XIAP↓, Grp78↑, α-CP1↓, Bcl-2↓, caspase-8↑, procaspase-3-9↓, pro-TNFα↑, p53↓, caspase-9↓, DeltaPsim↓, ERK↓, p38↑, MAPK↑, JNK↑
Oridonin induces growth inhibition and apoptosis of a variety of human cancer cells
PC-SPES is an eight herbal mixture that was shown to have activity against prostate cancer. Recently, we purified oridonin from Rabdosia rubescens, one component of PC-SPES, by high performance liquid chromatography (HPLC). The ability of oridonin to inhibit the proliferation of cancer cells was examined by MTT assay. Oridonin effectively inhibited the proliferation of a wide variety of cancer cells including those from prostate (LNCaP, DU145, PC3), breast (MCF-7, MDA-MB231), non-small cell lung (NSCL) (NCI-H520, NCI-H460, NCI-H1299) cancers, acute promyelocytic leukemia (NB4), and glioblastoma multiforme (U118, U138) with ED50s ranging from 1.8 to 7.5 µg/ml. TUNEL assay and cell cycle analysis showed that oridonin induced apoptosis and G0/G1 cell cycle arrest in LNCaP prostate cancer cells. In addition, expression of p21waf1 was induced in LNCaP and NCI-H520 cells in a p53-dependent manner. Interestingly, when p53 was suppressed by over-expression of E6 from human papilloma virus type 16 (HPV-16), these cells lost their sensitivity to oridonin-induced growth inhibition and apoptosis. Taken together, oridonin inhibited the proliferation of cancer cells via apoptosis and cell cycle arrest with p53 playing a central role in several cancer types which express the wild-type p53 gene. Oridonin may be a novel, adjunctive therapy for a large variety of malignancies and probably represents one of the major, active components of PC-SPES.
induces apoptosis via altering expression of Bcl-2/Bax and activating caspase-3/ICAD pathway.
AIM: To study the mechanisms by which oridonin inhibited HeLa cell growth in vitro. METHODS: Viability of oridonin-induced HeLa cells was measured by MTT assay. Apoptotic cells with condensed nuclei were visualized by phase contrast microscopy. Nucleosomal DNA fragmentation was assayed by agarose gel electrophoresis. Caspase activity was assayed using fluorometric protease assay. ICAD, Bcl-2, and Bax proteins expression were detected by Western blot analysis. RESULTS: Oridonin induced oligonucleosomal fragmentation of DNA and increased caspase-3 activity, on the other hand, reduced the expression of inhibitor of caspase-3-activated DNase (ICAD), a caspase-3 substrate, at 12 h in HeLa cells. Oridonin-induced DNA fragmentation, caspase-3 activation and down-regulation of ICAD expression were effectively inhibited by a caspase-3 inhibitor, z-DEVD-fmk (z-Asp-Glu-Val-Asp-fmk). However, pretreatment with an inhibitor of poly (ADP-ribose) polymerase (PARP), 3, 4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (DPQ), did not suppress oridonin-induced HeLa cell death. In addition, oridonin-induced apoptosis was associated with an increase in the expression of the apoptosis inducer Bax, and a significant reduction in expression of the apoptosis suppressor Bcl-2 in mitochondria. CONCLUSION: Oridonin induces HeLa cells apoptosis by altering balance of Bcl-2 and Bax protein expression and activation of caspase-3/ICAD pathway.
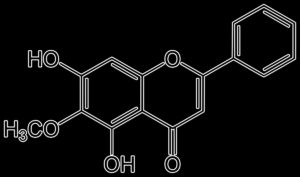
Oroxylin A
inhibits adipogenesis and induces apoptosis in 3T3-L1 cells
Oroxylin A (OA) is a flavonoid found in Oroxylum indicum, a medicinal plant with multiple biological activities. This study was taken up to investigate the effect of OA, on adipogenesis, lipolysis and apoptosis in3T3 L1cells. Pre-adipocytes were treated with 10–40 μM OA on various days of adipogenesis treatment schedule. Mature adipocytes were treated with OA for lipolysis and apoptosis studies. In maturing pre-adipocytes, 10 μM OA suppressed intracellular lipid accumulation by 42.19% which was confirmed by lipidTox imaging of cells. In addition, OA decreased the nuclear translocation of PPARγ and mRNA expression of its downstream genes (FAS and LPL) along with adiponectin secretion. In mature adipocytes, 40 μM of OA decreased cell viability by 30% of control. Annexin V/PI staining showed induction of apoptosis which was further confirmed by enhanced levels of pro-apoptotic proteins Bax, cyt c, AIF and chromatin condensation. OA enhanced TNF-αsecretion, lipolysis and decreased Akt phosphorylation in mature adipocytes. Findings suggest that OA possibly exerts its anti-obesity effect by affecting adipocyte life cycle at critical points of differentiation and maturity. When we compared the potency of OA with non-methoxylated flavonoids morin, naringenin and kaempferol on adipocyte life cycle OA was far more potent. Thus, study clearly indicates a new role for oroxylin A as regulator of adipocyte life cycle. In addition, study also suggested a specific role of methoxylated group in exerting lipolysis and cytotoxic effects in mature adipocytes.
O-vanillin
(from Vanilla planifolia aka “Vanilla Bean” pineoside, eugenol and safrole)
Key Function:
Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro
Curcumin and o-Vanillin cleared senescent intervertebral disc (IVD) cells and reduced the senescence-associated secretory phenotype (SASP) associated with inflammation and back pain. Cells from degenerate and non-mildly-degenerate human IVD were obtained from organ donors and from patients undergoing surgery for low back pain. Gene expression of senescence and SASP markers was evaluated by RT-qPCR in isolated cells, and protein expression of senescence, proliferation, and apoptotic markers was evaluated by immunocytochemistry (ICC). The expression levels of SASP factors were evaluated by enzyme-linked immunosorbent assay (ELISA). Matrix synthesis was verified with safranin-O staining and the Dimethyl-Methylene Blue Assay for proteoglycan content. Western blotting and ICC were used to determine the molecular pathways targeted by the drugs. We found a 40% higher level of senescent cells in degenerate compared to non-mildly-degenerate discs from unrelated individuals and a 10% higher level in degenerate compared to non-mildly-degenerate discs from the same individual. Higher levels of senescence were associated with increased SASP. Both drugs cleared senescent cells, and treatment increased the number of proliferating as well as apoptotic cells in cultures from degenerate IVDs. The expression of SASP factors was decreased, and matrix synthesis increased following treatment. These effects were mediated through the Nrf2 and NFkB pathways.
Bax↑, Bcl2↓, mRNA↓ metalloproteinase-9↓, STAT3⊥, JNK↑, VEGF⊥, IL8⊥, ABCB5 transporter↓, Bcl-X(L)↓, survivin↓, cyclin D1↓, IL8↓ matrix metalloproteinase 9↓, Akt phosphorylation↓, NF-κB↓, p65/NF-κB↓, Ki67↓, p21↑, antioxidant N-acetyl-L-cystein⊥, glutathione S-transferase↓ STAT3⊥, JAK⊥, tBid↑ of caspase-3/8/9↑, poly (ADP-ribose) polymerase⊥, p-ERK↑, p-p38↑, p38 and SAPK/JNK↑, PKC-alpha⊥, procaspase-3↓, p65↓, VEGF⊥, IL6 mRNA⊥, IkappaB-alpha↑, p53↑, ROS↑, JNK↑, Bid↑
proapoptotic CASPASE-8 ACTIVATOR
Parthenolide is a sesquiterpene lactone responsible for the bioactivities of Feverfew. Besides its potent anti-inflammatory effect, this compound has recently been reported to induce apoptosis in cancer cells, possibly through mitochondrial dysfunction. In the present study, we attempted to examine parthenolide-mediated cell death signaling pathway by focusing on the involvement of Bcl-2 familymembers. Using a human colorectal cancer cell line COLO205, we first demonstrated that parthenolide acted through the cell death receptor pathway to activate caspase 8.Following caspase 8 activation, Bid, a proapoptotic Bcl-2 member, was cleaved and this cleavage then triggered Bax conformational changes and Bax translocation from cytosol to mitochondrial membrane. Meanwhile, another proapoptotic protein, Bak, was up-regulated and oligomerized on the mitochondrial membrane. All these alterations were found to be prerequisite for the subsequent release of proapopototic mitochondrial proteins, including cytochrome c and Samc, in parthenolide-treated cells. Moreover, selective inhibition of caspase 8 activity by a synthetic caspase inhibitor (IETD-FMK) or overexpression of a viral protein (CrmA) suppressed the cleavage of Bid, conformational changes of Bax, cytochrome crelease, and apoptosis. Therefore, the proapoptotic Bcl-2 family members are important mediators relaying the cell death signaling elicited by parthenolide from caspase 8 to downstream effector caspases such as caspase 3, and eventually to cell death.

Patrinia scabiosaefolia
downregulates anti-apoptotic Bcl-2/Bcl-XL expression, and induces apoptosis in human breast carcinoma MCF-7 cells independent of caspase-9 activation
Patrinia scabiosaefolia Fisch. is a Chinese medicinal herb used traditionally for treating intestinal carbuncle. Although Patrinia scabiosaefolia has also been suggested for cancer therapy, there has not been any scientific evidence supporting this application. In this study, a panel of human cancer cells, including breast carcinoma MCF-7; hepatocellular carcinoma HepG2; skin melanoma A375; lung carcinoma A549 and prostate adenocarcinoma PC-3, were treated in vitro with ethyl acetate extract of Patrinia scabiosaefolia (EAE-PS) for 48 h. Results from MTT study showed that MCF-7 was the most responsive (IC50 = 112.3 μg/ml) while PC-3 was the most resistant (IC50 = 348.7 μg/ml) one to cell growth inhibition. DNA flow cytometry demonstrated that EAE-PS induced apoptosis in the resistant MCF-7 cells by 14.5-fold of the control level after 36 h of treatment. Immunoblot studies further illustrated that although EAE-PS downregulated the anti-apoptotic Bcl-2/Bcl-XL expression in breast cancer cells, the induced apoptosis could not be prevented by the caspase-9 inhibitor (Z-LEHD-FMK). All these results suggest that EAE-PS retards MCF-7 cell growth by activating the caspase-independent mitochondrial cell death pathway. Results from this study support future research and development of the bioactive ingredients from Patrinia scabiosaefolia as anticancer agents, especially against those apoptosis-resistant cancers with deregulated Bcl-2/Bcl-XL expression
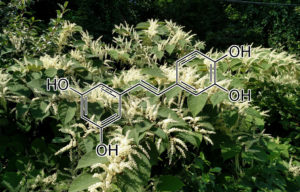
piceatannol
NOXA ACTIVATOR / XIAP (X-linked inhibitor of apoptosis protein) AKT & MTOR INHIBITOR
Resistance to cisplatin (CDDP) in ovarian cancer (OVCA) arises from the dysregulation of tumor suppressors and survival signals. During genotoxic challenge, these factors can be influenced by sec- ondary agents that facilitate the induction of apoptosis. Piceatan- nol is a natural metabolite of the stilbene resveratrol found in grapes and is converted from its parent compound by the enzyme CYP1BA1 p450. It has been hypothesized to exert specific effects against various cellular targets; however, its ability to influence CDDP resistance in cancer cells has not been investigated to date. Here, we show that piceatannol is a potent enhancer of CDDP sensitivity in OVCA, and this effect is achieved through the modulation of several major determinants of chemoresistance. Picea- tannol enhances p53-mediated expression of the pro-apoptotic protein NOXA, increases XIAP degradation via the ubiquitin-proteasome pathway, and enhances caspase-3 activation. This response is associated with an increase in Drp1-de- pendent mitochondrial fission, leading to more effective induction of apoptosis. In vivo studies using a mouse model of OVCA reveal that a number of these changes occur in association with a greater overall reduction in tumor weight when mice are treated with both piceatannol and CDDP, in comparison to treatment with either agent alone. Taken together, these findings demonstrate the potential application of piceatannol to enhance CDDP sensitivity in OVCA, and it acts on p53, XIAP, and mitochondrial fission.
In this study, we tested the hypothesis that piceatannol controls proliferation of both androgen-dependent (AD) and rogen-independent (AI) CaP cells by targeting the expression of mTOR. We also determined whether piceatannol disrupts the mTOR signaling pathway in CaP cells by eliciting changes in mTOR and its upstream and downstream effector proteins: mTOR, protein kinase AKT, initiation factor eIF- 4E regulatory binding protein eIF-4E-BP1, and ribosomal protein p70 S6 kinase. We found that piceatannol suppressed AD and AI CaP cell proliferation and that its growth inhibitory activity was accompanied by reduced expression of mTOR and its key effectors AKT and eIF4EBP-1.

Phloretin
(a dihydrochalcone, a type of natural phenol found in apple tree leaves and the Manchurian apricot.)
Phloretin as Senolytic
In vitro glucose metabolism was blocked by glucose transport inhibitors phloretin or cytochalasin B or by 2-deoxy-D-glucose. Glucose-free medium was supplemented with D-Glucose. The lactate dehydrogenase enzyme was inhibited by sodium oxamate. The electron transport chain was interrupted with antimycin A . AMPK was inhibited by Compound C (Sigma, 10mM). The carnitine palmitoyltransferase I inhibitor etomoxir (Sigma, 100M) was used to block fatty acid oxidation. In LC lysosomal V- ATPases were inhibited by Bafilomycin A1 or Concanamycin A. Lysosomal proteases were blocked using an inhibitor cocktail composed of pepstatin A, ZPAD (N-CBZ-l-phenylalanyl-l-alanine-diazomethylketone), and E64d. The final inhibitor concentrations were determined for each cell type by dose-response experiments with serial dilutions, so that the selected concentration specifically killed senescent cells.
These cells were also shown to be sensitive to another blocker of glucose transporters, cytochalasin B, thus suggesting that the mechanism by which phloretin induces cell death in senescent cells is related to their increased metabolic requirement.
upregulatES Nrf2 defensive pathway
Taken together, phloretin exhibited neuroprotective effects in cerebral ischemia/reperfusion, and the mechanisms are associated with oxidative stress inhibition and Nrf2 defense pathway activation.
Phloretin exhibits an anticancer effect by regulating expression of apoptotic pathways and matrix metalloproteinases.
We further confirmed that phloretin dose-dependently suppressed the expression of Bcl-2, increased the protein expression of cleaved-caspase-3 and -9, and deregulated the expression of matrix metalloproteinases (MMP)-2 and -9 on gene and protein levels.
Phloretin as Toxic Compound in Cancer
Phloretin is known to inhibit glucose transporter (GLUT) 2, a process which results in the induction of apoptosis in cells with high metabolic requirement, as shown in human liver cancer cells HepG2 treated with 200 μM phloretin [258]. At the dose of 10 mg/kg, phloretin was found to exert antitumor effects in immune deficiency mice carrying a HepG2 xenograft [258].
Moreover, phloretin was shown to induce apoptosis of non-small-cell lung cancer (NSCLC) cell line A549, Calu-1, H838, and H520 (IC50 approx. from 50 to 100 μM) through deregulation of Bcl-2 [259] and other ROS-related pathways, such as P38 MAPKand JNK1/2 [260] which are related to the rise of ROS. Interestingly, the anticancer effects were enhanced in presence of cisplatin, which suggest a potential in adjuvant cancer therapy. Similar proapoptotic effects, associated with increased ROS and ROS-related pathways, were observed after treatment with very high (200–300 μM) concentrations of phloretin [261].
cytoprotective effects
Phloretin at 2.5 and 5µM concentrations were able to protect the cells from ATO toxicity via protecting mitochondria through its antioxidant potential. The present investigation based on mitochondria reveals the probability of cardioprotective potential of phloretin for the cancer patients on ATO chemotherapy.
Phloretin induces apoptosis via JNK1/2 and p38 MAPK pathways
Previous studies have suggested that MAPKs can be induced by various compounds and are involved in cell death in NSCLC A549 cells (30,31). The MAPK family includes three kinase members, including c-Jun NH2-terminal protein kinase/stress activated protein kinases (JNK/SAPKs), P38 MAPK, and extracellular signal-regulated kinase (ERK). Previous results tempted us to ask whether the tumor-suppressing effect of Ph relied on the presence of the P38 MAPK signaling system in A549 cells. To answer this question, we further investigated activation of the MAPK family proteins in Ph-treated A549 cells. The results showed that the phosphorylation of ERK1/2, JNK1/2 and P38 MAPK was increased in Ph-treatment A549 cells in a dose-dependent manner with the total protein level remaining steady. However, treatment with JNK1/2 specific inhibitor (SP600125) or the P38 MAPK specific inhibitor (SB202190) effectively inhibited activation of caspase-3 and caspase-9 induced by Ph, whereas U0126 (an ERK1/2 inhibitor) showed no effect on Ph-induced caspase activation. These findings suggest that activation of JNK1/2 and P38 MAPK plays a critical role in Phloretin-induced apoptosis in NSCLC A549 cells.
MITOCHONDRIAL UNCOUPLER
Under all circumstances, phloretin lowered the transmembrane electrical potential difference in isolated mitochondria. We conclude that phloretin is both an uncoupler and an inhibitor of oxidative phosphorylation
Inhibition of Glucose Uptake by Phloretin
Enhanced glycolysis, a feature of many rapidly growing solid tumors, is associated with elevated expression of glucose transporter proteins (GLUT) and other glycolytic enzymes in neoplastic cells. Treatment with phloretin sensitized human colon cancer (SW620) and leukemia (K562) cells to daunorubicin by reducing the glucose uptake by these cells [63]. Lin et al. [25] reported that elevated levels of GLUT2 mRNA were detected in human colon cancer (COLO 205 and HT29) cells as well as in colorectal cancer tissues. Treatment with phloretin attenuated GLUT2 mRNA and protein expression by blocking hepatocyte nuclear factor 6 (HNF6), which is a transcription factor regulating GLUT2 expression. When compared to normal hepatocytes, the levels of GLUT2 mRNA were found to be five times greater in human hepatoma (HepG2) cells. Phloretin re-sensitized these cells to paclitaxel and induced apoptosis via caspase activation by attenuating GLUT2 expression. Thus, the growth of HepG2 cells xenograft tumors in nude mice markedly diminished by co-treatment with phloretin and paclitaxel, as compared to treatment with paclitaxel alone [15]. Another study has shown that GLUT2 expression is elevated in HepG2 cells and that the siRNA-mediated silencing of GLUT2 induced apoptosis in these cells [17]. While co-treatment of these cells with glucose uptake inhibitor cytochalasin B enhanced phloretin-induced apoptosis, the effect of phloretin was reversed by pretreatment with glucose. These findings suggest that induction of apoptosis in HepG2 cells by phloretin is mediated through inhibition of GLUT2 expression. Likewise, phloretin inhibited the proliferation of rat mammary adenocarcinoma and Fischer bladder cell carcinoma cells and arrested the growth of xenograft tumors of these cells in mice by blocking glucose transmembrane transport [64].
Phloretin as a Potential Cancer Immunotherapy Agent
The emergence of immunotherapies in recent years is considered a major breakthrough in the development of cancer therapeutics. Reinvigoration of exhausted T cells by blocking immune checkpoint markers, such as cytotoxic T cells antigen-4 (CTLA4), programmed cell death 1 (PD-1) and PD-1 ligand (PDL-1), have recently received Food and Drug Administration approval for the treatment of melanoma, renal cell carcinoma, and non-small cells lung cancer [65]. Although substantial progress has been made in developing cancer immunotherapies, the potential of natural compounds in boosting antitumor immunity is yet to be investigated. A recent study by Zhu et al. [66] reported that phloretin can enhance the tumoricidal effect of γδ T cells on human colon cancer (SW-1116) cells, possibly by stimulating the proliferation of γδ T cells. In another study, phloretin potentiated the anti-cancer effects of intratumorally administered recombinant heat shock protein-70 (HSP70) in B16 mouse melanoma cells, through enhancement of tumor cell sensitivity to cytotoxic lymphocytes by 16%–18% as compared to a treatment with HSP-70 alone. The combined treatment with phloretin and recombinant HSP-70 resulted in B16 melanoma cells xenograft tumor growth and increased the life span of tumor-bearing mice through the activation of innate and adaptive immunity. Authors also demonstrated that rHSP70 was more active in induction of CD8+ cell function and interferon-γ (γIFN) production, but phloretin induced CD56+ cell response [42].
Antiproliferative and Apoptosis Inducing Effects of Phloretin
There is accumulating evidence suggesting that phloretin arrests the proliferation of tumor cells and induces apoptosis in various human cancer cells, including those of the skin, colon, breast and prostate in culture. The underlying mechanisms of the antiproliferative effects of phloretin appears to be the interference with cell cycle progression via modulation of cyclins and induction of mitochondria-dependent programmed cell death. Park et al. [14] reported that the induction of apoptosis in human colon cancer (HT-29) cells upon treatment with phloretin was associated with increased expression of Baxand release of cytochrome c and Smac/DIABLO in the cytosol, thereby resulting in the cleavage of caspase-8, -9, -7, and -3 and poly(ADP-ribose) polymerase (PARP) [14]. A recent study demonstrated that phloretin induced apoptosis selectively in human gastric cancer cell lines (MGC80-3, BGC-823, SGC-7901, SNU-1, SNU-5, RF-1 and AGS), with IC50 values ranging from 8 to 32 µM without affecting the growth of normal gastric epithelial cells [39]. Incubation of AGS cells with phloretin reduced the colony formation in a concentration-dependent manner by arresting cell cycle at the G2/M phase. Moreover, the induction of apoptosis in AGS cells by phloretin was associated with the increase in Bax expression, inhibition of Bcl-2 levelsand decrease in the phosphorylation of c-Jun N-terminal kinase (JNK) and p38 MAP kinase [39]. In contrast, the activation of JNK and p38 MAP kinase has been attributed to phloretin-induced caspase-3 cleavage in H-ras-transformed human mammary epithelial cells (H-ras-MCF-10A) [55]. These authors also reported that phloretin attenuated the proliferation of H-ras-MCF-10A cells in a concentration-dependent manner and induced apoptosis via upregulation of p53 and Bax and the cleavage of PARP [55]. Although the induction of apoptosis in B16 melanoma cells by phloretin was associated with the elevated expression of Bax and caspase activation, the compound did not affect the expression of p53 or that of anti-apoptotic proteins Bcl-2 or Bcl-xl [56]. However, according to a recent study phloretin inhibited the expression of Bcl-2 and X-linked inhibitor of apoptosis (XIAP), and activated p53 as the mechanism of apoptosis induction in human esophageal cancer (EC-109) cells [40]. Phloretin arrested cell cycle at the G0/G1 phase and reduced proliferation of human breast cancer (MDA-MB-231) cells in a p53 mutant-dependent manner as evidenced by pre-incubating cells with a p53-specific dominant-negative expression vector [28]. Thus, the effects of phloretin on MAP kinase activation, Bcl-2 expression and p53 induction may be a cell-type specific phenomena.
Kobori et al. reported that phloretin caused apoptosis in human leukemia (HL60) cells, which was associated with decreased protein kinase C (PKC) activity [57]. Various chalcone compounds have been shown to prevent prostate cancer [58]. Phloretin, being a dihydrochalcone, significantly enhanced TNFα-related apoptosis-inducing ligand (TRAIL)-induced apoptosis and cytotoxicity in human prostate cancer (LNCaP) cells [58].
Phloretin attenuated growth and induced apoptosis in a number of lung cancer cells in culture. Treatment of human lung cancer (A549, Calu-1, H838 and H520) cells with phloretin resulted in reduced expression of anti-apoptotic protein Bcl-2 and elevation in the protein expression of cleaved-caspase-3 and -9 [41]. The induction of apoptosis in A549 cells by phloretin was associated with increased levels of Bax, cleavage of caspase-3 and -9, and PARP, and decreased Bcl-2 expression. Mechanistically, phloretin-induced caspase activation was mediated through the induction of p38 MAP kinase and JNK1/2 phosphorylation, as inhibition of these kinases by using specific inhibitors significantly abolished the phloretin-induced caspase activation [18].
The anti-proliferative effects of phloretin are also mediated through the modulation of cyclins and cyclin-dependent kinases (CDKs). According to a recent study, phloretin induced cell cycle arrest at the G0/G1 phase in human glioblastoma cells, at least in part, by increasing the expression of p27 and dampening that of CDK-2, -4, and -6, and cyclin-D, and -E. In addition, the compound suppressed signaling through the PI3K/AKT/mTOR cascades, resulting in reduced cell proliferation. The study also demonstrated that phloretin triggered the mitochondria-mediated cell death via generation of ROS, up-regulation of Bax, Bak and c-PARP, and the inhibition of Bcl-2[59].
Anti-Inflammatory and Anti-Cancer Effects of Phloretin
The biochemical mechanisms of anti-inflammatory and anti-cancer effects of phloretin have been widely investigated (). Phloretin has been shown to scavenge peroxynitrite radicals and inhibit lipid peroxidation, largely due to the presence of 2,6-dihydroxyacetone moiety as the 130 pharmacophore [29]. In another study, Nakamura et al. [12] demonstrated that the presence of a hydroxyl group at 2′-position of all dihydrochalcones, including phloretin, is an essential pharmacophore for the radical scavenging and lipid peroxidation activities. The bond dissociation enthalpies (BDEs) for hydroxyl (-OH) moieties (an indicator of antioxidant activity) of phloretin has recently been examined in a computational modeling study. The authors demonstrated that compared to well-known antioxidants, the BDEs among the four OH groups for phloretin were found to be the lowest, indicating the antioxidant potential of the compound [30]. Phloretin prevented oxidative DNA damage by restoring cellular glutathione (GSH) levels in human colon cancer (Caco-2 and HT-29) cells [31]. Likewise, the elevated cellular GSH level in phloretin-treated rat hepatocytes has been attributed to the induction of γ-glutamyl cysteine ligase (GCL), a rate limiting enzyme in GSH synthesis, via activation of a redox-regulated transcription factor, nuclear factor-erythroid related factor-2 (Nrf2) signaling [32]. These authors also demonstrated that phloretin induced the expression of another Nrf-2-regulated antioxidant enzyme, hemeoxygenase-1 (HO-1), in rat hepatocytes, via the activation of extracellular signal regulated kinase (ERK). By virtue of its antioxidative properties, phloretin attenuates the activation of exogenously exposed carcinogens as well as endogenous accumulation of damaged DNA or proteins, and inhibits the aberrant activation of various kinases and transcription factors involved in inflammatory signaling pathways. The following section will focus on detailing the molecular targets of phloretin as an anti-cancer agent ().
Inhibitory Effects of Phloretin on Inflammatory Markers
While inflammatory tissue damage is an initial trigger for tumor development, persistent intra-tumoral inflammation contributes to the invasion and metastasis of tumors. The key mediators of inflammation involved in neoplastic transformation of cells include, but are not limited to, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), and the cellular products catalyzed these enzymes, such as nitric oxide (NO) and prostaglandins (PGs) [22]. Studies have shown that mice harboring transgenic overexpression of cox-2 in the stomach [44] and skin [45] are highly prone to develop tumors in these organs, whereas genetic ablation of cox-2 protects mice against gastro-intestinal [46] and skin carcinogenesis [47]. The elevated levels of iNOS and its catabolic product NO have promoted dextran sulfate sodium-induced polyp formation in the colon of APCmin+ mice as compared to wild type mice [48]. The role of iNOS in skin papillomas has been evident from the inhibition of chemically induced mouse skin tumor development upon treatment with aminoguanidine, which is an iNOS inhibitor [49]. A large number of cytokines, for example, interleukins (IL) and tumor necrosis factor-α (TNFα), and chemokines also act as inflammatory mediators and participate in the tumorigenic process. The burst of inflammatory mediators requires inappropriate amplification of intracellular signaling cascades comprising various kinases and transcription factors. Aberrant activation of MAP kinases, Janus-activated kinase (JAK), and protein kinase B (PKB)/Akt have been implicated in precipitating inflammation and cancer. These upstream kinases transmit activating signals to a variety of redox-sensitive transcription factors, such as nuclear factor-kappaB (NF-κB), activator protein-1 (AP-1), signal transducer and activator of transcription (STAT), which leads to transactivation of genes encoding proteins involved in inflammation and oncogenesis [8,50].
Phloretin exerts anti-cancer effects by curbing inflammatory responses. For instance, phloretin markedly diminished the expression of pro-inflammatory genes by repressing the activation of NF-κB-, IL-8-, and STAT1-dependent signal transduction in human colon cancer (DLD1) cells and other immune cells (T84, MonoMac6, Jurkat) in a concentration-dependent manner [51]. The compound also inhibited TNFα-induced inflammatory responses in human colonic epithelial cells [24]. Shin et al. [19] reported that topical application of phloretin reduced phorbol ester-induced COX-2 expression and skin inflammation in mice by blocking the activation of NF-κB and ERK-mediated signaling. Moreover, pretreatment with phloretin diminished the production of various inflammatory markers, such as NO, PGE2, IL-6, and TNFα, and attenuated the expression of iNOS and COX-2 in lipopolysaccharide (LPS)-stimulated murine macrophage RAW264.7 cells. According to this study, phloretin blocked nuclear localization of the p65 protein, a component of NF-κB, and negated the phosphorylation in MAP kinases [16]. Phloretin reduced the expression of COX-2 and intracellular adhesion molecule -1 (ICAM-1), and the production of IL-6 in human lung epithelial (A549) cells stimulated with IL-1β by blocking the activation of NF-κB via downregulation of Akt and MAP kinases phosphorylation [35]. Likewise, the secretion of various cytokines and chemokines, such as IL-6, IL-8, and monocyte chemoattractant protein-1(MCP1), and the reduced expression of ICAM-1 in TNF-α-stimulated HaCaT keratinocytes by phloretin, have been attributed to the inactivation of NF-κB and MAP kinases [36]. Huang et al. recently reported that treatment with phloretin attenuated the gene expression of a variety of inflammatory markers, such as COX-2, iNOS, CCL5, MCP1, and ICAM-1 in LPS-treated mouse lung tissue [22,34,36,52,53].
Phloroglucinol
Phosphatidylcholine
induces apoptosis of 3T3-L1 adipocytes
Background Phosphatidylcholine (PPC) formulation is used for lipolytic injection, even though its mechanism of action is not well understood.
Methods The viability of 3T3-L1 pre-adipocytes and differentiated 3T3-L1 cells was measured after treatment of PPC alone, its vehicle sodium deoxycholate (SD), and a PPC formulation. Western blot analysis was performed to examine PPC-induced signaling pathways.
Results PPC, SD, and PPC formulation significantly decreased 3T3-L1 cell viability in a concentration-dependent manner. PPC alone was not cytotoxic to CCD-25Sk human fibroblasts at concentrations <1 mg/ml, whereas SD and PPC formulation were cytotoxic. Western blot analysis demonstrated that PPC alone led to the phosphorylation of the stress signaling proteins, such as p38 mitogen-activated protein kinase and c-Jun N-terminal kinase, and activated caspase-9, -8, -3 as well as cleavage of poly(ADP-ribose) polymerase. However, SD did not activate the apoptotic pathways. Instead, SD and PPC formulation induced cell membrane lysis, which may lead to necrosis of cells.
Conclusions PPC results in apoptosis of 3T3-L1 cells.

Phyllanthus emblica fruit extract
attenuates lipid metabolism in 3T3-L1 adipocytes via activating apoptosis mediated cell death
Background
Phyllanthus emblica L. (Indian gooseberry) is widely used in the Ayurveda for thousands of years to treat health complications including disorders of the immune system, diabetes, and obesity.
Purpose
For the first time, our study aims to demonstrate the molecular mechanisms of the fruit extract of Phyllanthus emblica (PEFE) involved in the promotion of fat cell apoptosis and alleviation of adipogenesis.
Methods
The active constituents from PEFE were identified using high performance liquid chromatography-mass spectrometry (HPLC-MS). We carried out the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay to evaluate the cytotoxic effects of PEFE using 3T3-L1 pre-adipocytes. The colonogenic assay was carried out to determine the inhibitory effect of 3T3-L1 adipocytes after PEFE treatment. In addition, inhibition of pancreatic lipase activity was performed and the lipolytic activity of PEFE and digallic acid was compared with the well-known standard drug orlistat. Besides, the molecular interaction and ligand optimization between digallic and adipogenesis/apoptosis markers were also carried out. Furthermore, to confirm fat cell apoptosis we have used several detection methods that includes Hoechst staining, PI staining, Oil staining and qPCR respectively.
Results
Digallic acid was identified as a major component in the PEFE. The IC50 values of digallic acid and PEFE were found to be 3.82 µg/ml and 21.85 µg/ml respectively. PEFE and digallic acid showed significant anti-lipolytic activity compared to the standard drug orlistat. In the mature adipocytes, PEFE significantly decreased triglyceride accumulation by downregulating adiponectin, PPARγ, cEBPα, and FABP4respectively. We further analyzed the expression of apoptosis related genes upon PEFE treatment. Apoptotic process initiated through upregulation of BAX and downregulation of BCL2 resulting in an increased caspase-3 activity. In addition, we have also confirmed the apoptosis and DNA fragmentation in 3T3-L1 cells using Hoechst, PI and TUNEL assays.
Conclusion
PEFE negatively regulates adipogenesis by initiating fat cell apoptosis and therefore it can be considered as a potential herbal medicinal product for treating obesity.

PiperlonguminE (Piper longum)
(from Piper longum L. aka “Long Pepper”)
Discovery of piperlongumine as a potential novel lead for the development of senolytic agents
Accumulating evidence indicates that senescent cells play an important role in many age-associated diseases. The pharmacological depletion of senescent cells (SCs) with a “senolytic agent”, a small molecule that selectively kills SCs, is a potential novel therapeutic approach for these diseases. Recently, we discovered ABT-263, a potent and highly selective senolytic agent, by screening a library of rationally-selected compounds. With this screening approach, we also identified a second senolytic agent called piperlongumine (PL). PL is a natural product that is reported to have many pharmacological effects, including anti-tumor activity. We show here that PL preferentially killed senescent human WI-38 fibroblasts when senescence was induced by ionizing radiation, replicative exhaustion, or ectopic expression of the oncogene Ras. PL killed SCs by inducing apoptosis, and this process did not require the induction of reactive oxygen species. In addition, we found that PL synergistically killed SCs in combination with ABT-263, and initial structural modifications to PL identified analogs with improved potency and/or selectivity in inducing SC death. Overall, our studies demonstrate that PL is a novel lead for developing senolytic agents.
Piperlongumine, derived from the fruit of the Long pepper plant (Piper longum), comes from southern India and southeast Asia. It is known as a potent anti-inflammatory, anti-atherosclerotic and anti-tumor agent. Specifically,Piperlongumine suppresses the production of Tumor Necrosis factor-α and interleukin-6 and inhibits the activation of nuclear factor-κB (NF-κB) against proinflammatory responses. Importantly as a senolytic, it inactivates the phosphatidylinositol-3-kinase/protein kinase B (Akt)/mammalian target of rapamycin, mitogen-activated protein kinase 14/c-Jun N-terminal kinase, NF-κB, and signal transducer and activator of transcription 3 (STAT3) pathways.
Piperlongumine is a natural alkaloid isolated from the long pepper. It is a potent inducer of Nrf2 response and of its target genes including heme oxygenase-1 (HO-1) [50]. Interestingly, HO-1 has antitumor functions in cancer cells, but cytoprotective functions in normal cells.
Senolytic activity of piperlongumine analogues: Synthesis and biological evaluation
Selective clearance of senescent cells (SCs) has emerged as a potential therapeutic approach for age-related diseases, as well as chemotherapy- and radiotherapy-induced adverse effects. Through a cell-based phenotypic screening approach, we recently identified piperlongumine (PL), a dietary natural product, as a novel senolytic agent, referring to small molecules that can selectively kill SCs over normal or non-senescent cells. In an effort to establish the structure-senolytic activity relationships of PL analogues, we performed a series of structural modifications on the trimethoxyphenyl and the α,β-unsaturated δ-valerolactam rings of PL. We show that modifications on the trimethoxyphenyl ring are well tolerated, while the Michael acceptor on the lactam ring is critical for the senolytic activity. Replacing the endocyclic C2–C3 olefin with an exocyclic methylene at C2 render PL analogues 47–49with increased senolytic activity. These α-methylene containing analogues are also more potent than PL in inducing ROS production in WI-38 SCs. Similar to PL, 47–49reduce the protein levels of oxidation resistance 1 (OXR1), an important oxidative stress response protein that regulates the expression of a variety of antioxidantenzymes, in cells. This study represents a useful starting point toward the discovery of senolytic agents for therapeutic uses.
Piperlongumine promotes autophagy via inhibition of Akt/mTOR signalling and mediates cancer cell death
Background: The Akt/mammalian target of rapamycin (mTOR) signalling pathway serves as a critical regulator of cellular growth, proliferation and survival. Akt aberrant activation has been implicated in carcinogenesis and anticancer therapy resistance. Piperlongumine (PL), a natural alkaloid present in the fruit of the Long pepper, is known to exhibit notable anticancer effects. Here we investigate the impact of PL on Akt/mTOR signalling.
Methods: We examined Akt/mTOR signalling in cancer cells of various origins including prostate, kidney and breast after PL treatment. Furthermore, cell viability after concomitant treatment with PL and the autophagy inhibitor, Chloroquine (CQ) was assessed. We then examined the efficacy of in vivo combination treatment using a mouse xenograft tumour model.
Results: We demonstrate for the first time that PL effectively inhibits phosphorylation of Akt target proteins in all tested cells. Furthermore, the downregulation of Akt downstream signalling resulted in decrease of mTORC1 activity and autophagy stimulation. Using the autophagy inhibitor, CQ, the level of PL-induced cellular death was significantly increased. Moreover, concomitant treatment with PL and CQ demonstrated notable antitumour effect in a xenograft mouse model.
Conclusions: Our data provide novel therapeutic opportunities to mediate cancer cellular death using PL. As such, PL may afford a novel paradigm for both prevention and treatment of malignancy.
Synthesis of Piperlongumine Analogues and Discovery of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Activators as Potential Neuroprotective Agents
The cellular antioxidant system plays key roles in blocking or retarding the pathogenesis of adult neurodegenerative disorders as elevated oxidative stress has been implicated in the pathophysiology of such diseases. Molecules with the ability in enhancing the antioxidant defense thus are promising candidates as neuroprotective agents. We reported herein the synthesis of piperlongumine analogues and evaluation of their cytoprotection against hydrogen peroxide- and 6-hydroxydopamine-induced neuronal cell oxidative damage in the neuron-like PC12 cells. The structure–activity relationship was delineated after the cytotoxicity and protection screening. Two compounds (4 and 5) displayed low cytotoxicity and confer potent protection of PC12 cells from the oxidative injury via upregulation of a panel of cellular antioxidant molecules. Genetically silencing the transcription factor Nrf2, a master regulator of the cellular stress responses, suppresses the cytoprotection, indicating the critical involvement of Nrf2 for the cellular action of compounds 4 and 5 in PC12 cells.
Synthesis and Antileukemic Activities of Piperlongumine and HDAC Inhibitor Hybrids against Acute Myeloid Leukemia Cells
Synergistic-to-additive antileukemic interactions of piperlongumine (PL) and HDAC inhibitor (HDACi) SAHA (Vorinostat) provide a compelling rationale to construct PL-HDACi hybrids, such as 1–58, which recapitulated the synergism between the parental compounds in high-risk and chemoresistant AML cells. Both PL and HDACi components, either in combination or in hybrid molecules, are essential for inducing significant DNA damage and apoptosis. Introducing C2-chloro substituent to 1–58 yielded 3–35 with increased cytotoxicity but decreased selectivity in noncancerous MCF-10A cells; eliminating C7–C8 olefin of PL obtained 3–31/3–98 scaffolds which were still more active than PL or SAHA in AML and were well-tolerated by MCF-10A cells. The HDACi function was crucial for modulating expression of DNA repair and apoptosis-related proteins. Collectively, PL and SAHA hybrids are potent, multifunctional anti-AML agents, acting in part, by interfering cellular GSH defense, suppressing expression of DNA repair and pro-survival proteins, and inducing expression of pro-apoptotic proteins.
Piperlongumine (piplartine) as a lead compound for anticancer agents – Synthesis and properties of analogues
Highlights • Piperlongumine is a promising antitumor agent. • This article describes synthesis and anticancer properties of piperlongumine analogues. • Some of them have showed increased anticancer properties and more appropriate drug-like parameters. • General structure-activity relationship conclusions are drawn and directions for future research are indicated.
Piperlongumine, also known as piplartine, is an amide alkaloid of Piper longum L. (long piper), a medical plant known from Ayurvedic medicine. Although was discovered well over fifty years ago, its pharmacological properties have been uncovered in the past decade. In particular, piperlongumine has been most extensively studied as a potential anticancer agent. Piperlongumine has exhibited cytotoxicity against a broad spectrum of human cancer cell lines, as well as demonstrated antitumor activity in rodents. Piperlongumine has also been found to be a proapoptotic, anti-invasive, antiangiogenic agent and synergize with modern chemotherapeutic agents. Because of its clinical potential, several studies were undertaken to obtain piperlongumine analogues, which have exhibited more potent activity or more appropriate drug-like parameters. In this review, the synthesis of piperlongumine analogues and piperlongumine-based hybrid compounds, as well as their anticancer properties and the molecular basis for their activity are explored. General structure-activity relationship conclusions are drawn and directions for the future research are indicated.
Piperlongumine (piplartine) as a lead compound for anticancer agents – Synthesis a Piperlongumine Induces Apoptosis in Colorectal Cancer HCT 116 Cells Independent of Bax, p21 and p53 Status
Background/Aim: Colorectal cancer is a common type of cancer with reported resistance to treatment, in most cases due to loss of function of apoptotic and cell-cycle proteins. Piperlongumine (PPLGM) is a natural alkaloid isolated from Piper species, with promising anti-cancer properties. This study investigated whether PPLGM is able to induce cell death in colorectal carcinoma HCT 116 cells expressing wild-type or deficient in Bax, p21 or p53. Materials and Methods: PPLGM was extracted from roots of Piper tuberculatum. Cell viability was determined by reduction of 3-(4,5-dimethilthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and clonogenic assay. Cell death was evaluated by acridine orange/ethidium bromide staining and flow cytometry. Plasmid cleavage activity and circular dichroism DNA interaction were also analyzed. Results: PPLGM induced selective cell death in all cell lines (IC50 range from 10.7 to 13.9 μM) with an increase in the number of late apoptotic cells and different profiles in cell-cycle distribution. Plasmid DNA analysis showed that PPLGM does not interact directly with DNA. Conclusion: This paper suggests that PPLGM may be a promising candidate in colorectal cancer therapy.
Piperlongumine induces apoptotic and autophagic death via activation of ROS-p38/JNK pathways
Aim: To investigate the effects of piperlongumine (PL), an anticancer alkaloid from long pepper plants, on the primary myeloid leukemia cells from patients and the mechanisms of action. Methods: Human BM samples were obtained from 9 patients with acute or chronic myeloid leukemias and 2 patients with myelodysplastic syndrome (MDS). Bone marrow mononuclear cells (BMMNCs) were isolated and cultured. Cell viability was determined using MTT assay, and apoptosis was examined with PI staining or flow cytometry. ROS levels in the cells were determined using DCFH-DA staining and flow cytometry. Expression of apoptotic and autophagic signaling proteins was analyzed using Western blotting. Results: PL inhibited the viability of BMMNCs from the patients with myeloid leukemias (with IC50 less than 20 μmol/L), but not that of BMMNCs from a patient with MDS. Furthermore, PL (10 and 20 μmol/L) induced apoptosis of BMMNCs from the patients with myeloid leukemias in a dose-dependent manner. PL markedly increased ROS levels in BMMNCs from the patients with myeloid leukemias, whereas pretreatment with the antioxidant N-acetyl-L-cysteine abolished PL-induced ROS accumulation and effectively reduced PL-induced cytotoxicity. Moreover, PL markedly increased the expression of the apoptotic proteins (Bax, Bcl-2 and caspase-3) and autophagic proteins (Beclin-1 and LC3B), and phosphorylation of p38 and JNK in BMMNCs from the patients with myeloid leukemias, whereas pretreatment with the specific p38 inhibitor SB203580 or the specific JNK inhibitor SP600125 partially reversed PL-induced ROS production, apoptotic/autophagic signaling activation and cytotoxicity. Conclusion: Piperlongumine induces apoptotic and autophagic death of the primary myeloid leukemia cells from patients via activation of ROS-p38/JNK pathways.
Inhibition of STAT-3 by piperlongumine induces anoikis, prevents tumor formation in pancreatic cancer in vitro and in vivo
Metastasis of pancreatic cancer is a major challenge leading to the failure of its therapy. Hence, novel strategies to prevent metastasis are of prime importance. Anoikis is an anchorage independent cell death. Resistance to anoikis is one of the key features of a metastatic cell. In the current study, we show induction of anoikis by piperlongumine, a component of black pepper, through inhibition of STAT-3. Treatment of various pancreatic cancer cell lines under low attachment conditions with piperlongumine reduced the anoikis resistance in a concentration-dependent manner. The decrease in anoikis resistance was associated with decrease in the expression of STAT-3 and its phosphorylation at Y705. Significant down-regulation of Bcl-2 and Mcl-1, which are under the transcriptional regulation of STAT-3, were also observed by piperlongumine treatment indicating the role of STAT-3 in anoikis. Concentration-dependent cleavage of poly (ADP-ribose) polymerase (PARP) was observed by piperlongumine treatment in these cells. IL-6 treatment not only increased the phophorylation of STAT-3 at Y05 but also abrogated the anoikis induced by piperlongumine in Panc-1 and HPAC cells. Silencing STAT-3 by shSTAT-3 sensitized BxPc-3 and Panc-1 cells to piperlongumine treatment. On the other hand, over-expression of STAT-3 in Panc-1 cells conferred resistance to anoikis induced by piperlongumine. STAT-3 -/- BxPc-3 cells were highly sensitive to anoikis upon piperlongumine treatment. Finally, our in vivo studies established that pancreatic cancer cells treated with piperlongumine in suspension condition lost their potential to form tumors in xenograft model. On the other hand, untreated cells in suspension conditions formed aggressive tumors. Moreover, cells treated with PL in suspension conditions completely failed to metastasize when injected intravenously in mice whereas the untreated cells extensively metastasized in lungs and liver. The presence of metastatic tumors was detected by bioluminescence imaging as well as immunohistochemistry. Taken together, our study suggests that the decrease in anoikis resistance induced by piperlongumine was mediated by STAT-3 inhibition which resulted in reduced tumorigenic as well as metastatic potential of pancreatic cancer cells. Notably, STAT-3 has been shown to be constitutively upregulated in pancreatic cancer rendering it resistant to chemotherapy and potential to metastasize.
Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer
Signal transducer and activator of transcription (STAT) 3 regulates many cardinal features of cancer including cancer cell growth, apoptosis resistance, DNA damage response, metastasis, immune escape, tumor angiogenesis, the Warburg effect and oncogene addiction and has been validated as a drug target for cancer therapy. Several strategies have been used to identify agents that target Stat3 in breast cancer but none has yet entered into clinical use. We used a high-throughput fluorescence microscopy search strategy to identify compounds in a drug-repositioning library (Prestwick library) that block ligand-induced nuclear translocation of Stat3 and identified piperlongumine (PL), a natural product isolated from the fruit of the pepper Piper longum. PL inhibited Stat3 nuclear translocation, inhibited ligand-induced and constitutive Stat3 phosphorylation, and modulated expression of multiple Stat3-regulated genes. Surface plasmon resonance assay revealed that PL directly inhibited binding of Stat3 to its phosphotyrosyl peptide ligand. Phosphoprotein antibody array analysis revealed that PL does not modulate kinases known to activate Stat3 such as Janus kinases, Src kinase family members or receptor tyrosine kinases. PL inhibited anchorage-independent and anchorage-dependent growth of multiple breast cancer cell lines having increased pStat3 or total Stat3, and induced apoptosis. PL also inhibited mammosphere formation by tumor cells from patient-derived xenografts. PL’s antitumorigenic function was causally linked to its Stat3-inhibitory effect. PL was non-toxic in mice up to a dose of 30 mg/kg/day for 14 days and caused regression of breast cancer cell line xenografts in nude mice. Thus, PL represents a promising new agent for rapid entry into the clinic for use in treating breast cancer, as well as other cancers in which Stat3 has a role.
Historical spice as a future drug: therapeutic potential of piperlongumine
Background: Spice and spice-derived compounds have been identified and explored for their health benefits since centuries. One of the spice long pepper has been traditionally used to treat chronic bronchitis, asthma, constipation, gonorrhea, paralysis of the tongue, diarrhea, cholera, malaria, viral hepatitis, respiratory infections, stomach ache, diseases of the spleen, cough, and tumors. Methods: In this review, the evidences for the chemopreventive and chemotherapeutic potential of piperlongumine have been described. Results: The active component piperlonguime has shown effective against various ailments including cancer, neurogenerative disease, arthritis, melanogenesis, lupus nephritis, and hyperlipidemic. These beneficial effects of piperlongumine is attributed to its ability to modulate several signaling molecules like reactive oxygen species, kinases, proteasome, proto-oncogenes, transcription factors, cell cycle, inflammatory molecules and cell growth and survival molecules. Piperlongumine also chemosensitizes to drugs resistant cancer cells. Conclusion: Overall the consumption of long peppers is therefore recommended for the prevention and treatment of various diseases including cancer, and thus piperlongumine may be a promising future candidate drug against cancer.
JNK signaling pathway is involved in piperlongumine-mediated apoptosis in human colorectal cancer HCT116 cells
Piperlongumine (PPLGM), an alkaloid isolated from the long pepper (Piper longum L.), can selectively trigger cancer cell death in colorectal cancer cells. The present study investigated whether the c-Jun NH2-terminal kinase (JNK) signaling pathway is involved in PPLGM-induced apoptosis in the human colorectal cancer HCT116 cell line. The results demonstrated that PPLGM reduced the cell viability and induced cell apoptosis in a time- and concentration-dependent manner, without a significant effect on cell cycle distribution. Meanwhile, treatment with 10 µM PPLGM resulted in JNK activationwithin 1 h, and a marked and sustained increase in c-Jun phosphorylation in the HCT116 cells. In addition, SP600125, a general inhibitor of JNK, inhibited PPLGM-induced apoptosis in the HCT116 cells by inhibiting PPLGM-induced c-Jun phosphorylation. Altogether, it can be concluded that the JNK signaling pathway, at least in part, is involved in PPLGM-mediated apoptosis in HCT116 cells.
Oxidation resistance 1 is a novel senolytic target
The selective depletion of senescent cells (SCs) by small molecules, termed senolytic agents, is a promising therapeutic approach for treating age‐related diseases and chemotherapy‐ and radiotherapy‐induced side effects. Piperlongumine (PL) was recently identified as a novel senolytic agent. However, its mechanism of action and molecular targets in SCs was unknown and thus was investigated. Specifically, we used a PL‐based chemical probe to pull‐down PL‐binding proteins from live cells and then mass spectrometry‐based proteomic analysis to identify potential molecular targets of PL in SCs. One prominent target was oxidation resistance 1 (OXR1), an important antioxidant protein that regulates the expression of a variety of antioxidant enzymes. We found that OXR1 was upregulated in senescent human WI38 fibroblasts. PL bound to OXR1 directly and induced its degradationthrough the ubiquitin‐proteasome system in an SC‐specific manner. The knockdown of OXR1expression by RNA interference significantly increased the production of reactive oxygen species in SCs in conjunction with the downregulation of antioxidant enzymes such as heme oxygenase 1, glutathione peroxidase 2, and catalase, but these effects were much less significant when OXR1 was knocked down in non‐SCs. More importantly, knocking down OXR1 selectively induced apoptosis in SCs and sensitized the cells to oxidative stress caused by hydrogen peroxide. These findings provide new insights into the mechanism by which SCs are highly resistant to oxidative stress and suggest that OXR1 is a novel senolytic target that can be further exploited for the development of new senolytic agents.
4.13.1. Piperlongumine as Toxic Compound in Cancer
Piperlongumine displays a high degree of selective toxicity to cancer cells. It has been identified as strong inhibitor (IC50 = 1.7 μM) of signal transducer and activator of transcription 3 (STAT3) by a recent high throughput drug-repository screening [245]. STAT3 is a validated drug target for cancer therapy and thus it is not surprising that piperlongumine was found to be able to induce apoptosis at low doses (IC50 from 0.16 up to 5.1 μM) in multiple breast cancer cell lines having increased STAT3. This proapoptotic activity is associated with the modulation of several antiapoptotic genes including Bcl-2, BcL-xL, survivin, X-linked inhibitor of apoptosis (XIAP), and cellular inhibitor of apoptosis proteins (cIAP). Alone and in combination with cisplatin, piperlongumine (2.5–15 μM) is able to dysregulate the oxidative stress response and kill head and neck cancer cells independently by their p53 mutational status [246] as well as a multitude of pancreatic, kidney, breast, lung, and pancreatic cell lines (Panc1, L3.6pL, A549, kidney, and SKBR3) [247]. In human oral squamous cell carcinoma, piperlongumine induces increased ROS and subsequent caspase-dependent apoptosis at 7.5–10 μM [248]. However, another study found no evidence of dose-response relationship between cellular ROS, induced by piperlongumine, and its cytotoxicity [249], thus suggesting the presence of different mechanisms related to induction of cell death.
Phloroglucinol
(Secondary metabolite found in a variety of marine organisms, especially brown “Ecklonia stolonifera, Eisenia bicyclis”, and bacteria “Pseudomonas fluorescens”)
BCL-2 & BCL-XL INHIBITOR
Anti-apoptotic Bcl-2 and Bcl-xL proteins are able to intercept diverse apoptotic signals, whereas pro-apoptotic proteins, such as Bax and Bad, can cause the release of apoptosis palpating factors into the cytoplasm (30,31). Our data showed that phloroglucinol induced a decrease in Bcl-2 and Bcl-xL expression and an increase in Bax and Bad expression.This alteration may trigger the release of cytochrome c and Apaf-1 into the cytosol and the cleavage of poly(ADP-ribose) polymerase (32). In this study, the release of apoptosis-promoting factors such as cytosolic cytochrome c and Apaf-1 was increased following phloroglucinol treatment. Therefore, these findings suggest that phloroglucinol induces apoptosis through mitochondrial dysfunction by modulating the protein levels of Bcl-2 family members. These findings may aid in the understanding of the mechanism underlying induction of apoptosis by phloroglucinol in carcinoma cells.
POMEGRANATE PEEL
TLR4 INHIBITOR
Inflammation is a primary physiological defense process of the biological system and protects against injuries caused by harmful stimuli such as pathogens and poisons (11). Natural products have long been recognized as an invaluable source of the most active components of medicines for treating and preventing various human diseases, including inflammatory diseases. Many studies have shown that phytochemicals such as polyphenols have anti-inflammatory effects. Pomegranate peel extracts have been focused for their richness and unique compositions in polyphenols. In our previous work, PPPs and their main polyphenol components (PC and EA) had been shown to significantly inhibit the expression of pro-inflammatory mediators and inflammatory cytokines in LPS-induced RAW264.7 macrophages. The molecular mechanism was associated with the inhibition of MAPKs activation. In the present work, we found that PPPs, PC, and EA significantly inhibited LPS-induced mRNA and protein expression of TLR4. TLRs are an integral part of the molecular mechanisms of inflammation processes. LPS activates macrophage and microglia by selectively stimulating toll-like receptor 4 (TLR4) and triggering NF-κB and MAPK pathways (32) specifically, and eventually results in the excess production of a large array of cytotoxic factors such as IL-1β, IL-6, TNF-α, NO, ROS, iNOS, and COX-2 (33, 34). This result indicated that the decreased expression of TLR4 could inhibit the initiation of intracellular signaling cascades, which subsequently suppressed the activation of NF-κB and pro-inflammatory mediators.
inhibits matrix metalloproteinase-1 (MMP-1)
Pomegranate as a cosmeceutical source: Pomegranate fractions promote proliferation and procollagen synthesis and inhibit matrix metalloproteinase-1 productionin human skin cells
Pomegranate (Punica granatum) is an ancient fruit with exceptionally rich ethnomedical applications. The peel (pericarp) is well regarded for its astringent properties; the seeds for conferring invulnerability in combat and stimulating beauty and fertility. Here, aqueous fractions prepared from the fruit’s peel and fermented juice and lipophilic fractions prepared from pomegranate seeds were examined for effects on human epidermal keratinocyte and human dermal fibroblast function. Pomegranate seed oil, but not aqueous extracts of fermented juice, peel or seed cake, was shown to stimulate keratinocyte proliferation in monolayer culture. In parallel, a mild thickening of the epidermis (without the loss of ordered differentiation) was observed in skin organ culture. The same pomegranate seed oil that stimulated keratinocyte proliferation was without effect on fibroblast function. In contrast, pomegranate peel extract (and to a lesser extent, both the fermented juice and seed cake extracts) stimulated type I procollagen synthesis and inhibited matrix metalloproteinase-1 (MMP-1; interstitial collagenase) production by dermal fibroblasts, but had no growth-supporting effect on keratinocytes. These results suggest heuristic potential of pomegranate fractions for facilitating skin repair in a polar manner, namely aqueous extracts (especially of pomegranate peel) promoting regeneration of dermis, and pomegranate seed oil promoting regeneration of epidermis.
inhibits activation of NF-kβ, MAPK, and downregulates COX2
Punica granatum (pomegranate) Pomegranate is a rich source of anthocyanins, ellagitannins and ellagic acid. In particular, ellagitannins and ellagic acid are potent polyphenolic compounds metabolized by gut microbiota to yield different types urolithins. There are two main forms of urolithin: urolithin-A and urolithin-B [86]. The anti-inflammatory role of ellagic acid is mainly due to its metabolite urolithin-A. This molecule inhibits activation of NF-kβ, MAPK, and downregulates COX2[87]. Dermatologic uses of ellagic acid are suggested by studies on human skin cells and hairless mice. Ellagic acid reduces in vitro UVB-induced expression of IL-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1), and TNF-α, as well as inhibits intercellular adhesion molecule-1 (ICAM-1)-UVR-dependent expression in keratinocytes. Ellagic acid blocks MMP secretion and collagen degradation also in UVR-exposed fibroblast, leading to less wrinkles and thickness [88, 89]. A 4-week oral supplementation with 100–200 mg/day of ellagic acid produced an inhibitory effect on a slight pigmentation in the human skin caused by UV irradiation [90]. Treatment of human reconstituted skin with pomegranate derived products reduced UVR-induced protein expression of c-Fos and phosphorylation of c-Jun. In addition, pomegranate derived products inhibited UVB-induced expressions of MMP-1, MMP-2, MMP-9, MMP-3, MMP-7, MMP-12 [91]. Finally, the antioxidant potential of ellagic acid was directly correlated with the transcriptional activation of Nrf2, which was followed by increased expression of HO-1 and SOD in human keratinocyte cells [92].

PRISTIMERIN
Pristimerin, a Naturally Occurring Triterpenoid, Exerts Potent Anticancer Effect in Colon Cancer Cells (BCL-2, BCL-XL INHIBITOR)
Pristimerin is a triterpene compound isolated from plant extracts that reportedly possesses antitumor, antioxidant, and anti-inflammatory activities. The current study was designed to evaluate the antitumor effects of pristimerin on human colon cancer cells. Treatment of the human colon cancer cells, HCT116 and SW480, with pristimerin led to a dose-dependent decrease in cell proliferation. Flow cytometry experiments showed that pristimerin increased cell apoptotic rate and decreased the mitochondrial membrane potential in HCT116 and SW480 cells. Western blot assay showed that pristimerin induced increased cleavage of caspase-3, -7, -8, and poly ADP ribose polymerase. Treatment with pristimerin also caused a marked decrease in the expression of Bcl-2 and Bcl-xL. Additionally, the levels of phosphorylated AKT and forkhead box O3a (FOXO3a) were decreased in pristimerin-treated colon cancer cells. Taken together, our study illustrated that pristimerin promoted apoptosis via the AKT/FOXO3a signaling pathway in colon cancer cells, elucidating that it might be considered as a potential agent for colon cancer therapy.
Pristimerin triggers AIF-dependent programmed necrosis in glioma cells via activation of JNK
•Pristimerin inhibits the growth of glioma cells in vitro and in vivo.•Pristimerin induced glioma cell death via AIF-dependent necrosis.•JNK activation leads to mitochondrial depolarization and nuclear accumulation of AIF.•ROS accounts for pristimerin-induced JNK activation.•Pristimerin has potential therapeutic effect in the treatment of glioma.
Programmed necrosis is established as a new form of programmed cell death and is emerging as a new strategy of treatment for cancers. Pristimerin is a natural chemical with anti-tumor effect despite the fact that its mechanism remains poorly understood. In this study, we used glioma cell lines and mice model of xenograft glioma to investigate the effect of pristimerin on glioma and its underlying mechanism. We found that pristimerin inhibited the viabilities of glioma cells in vitro and the growth of xenograft gliomas in vivo, which was accompanied by upregulation of JNK and phosphor-JNK, nuclear accumulation of AIF, and elevation in the ratio of Bax/Bcl-2. In vitro studies showed that pristimerin induced necrosis in glioma cells, as well as mitochondrial depolarization, overproduction of ROS and reduction of GSH. Ablation of AIF level with SiRNA mitigated pristimerin-induced nuclear accumulation of AIF and prevented necrosis in glioma cells. Moreover, pharmacological inhibition of JNK with SP600125 or knockdown of its level with SiRNA reversed mitochondrial depolarization attenuated the elevation of Bax/Bcl-2 and suppressed nuclear accumulation of AIF. Further, inhibition of ROS with NAC not only rescued glioma cell necrosis but also suppressed JNK activation, mitigated Bax/Bcl-2 ratio, maintained mitochondrial membrane potential, and inhibited AIF translocation into nucleus. Therefore, we demonstrated first in this study that pristimerin triggered AIF-dependent necroptosis in glioma cells via induction of mitochondrial dysfunction by activation of JNK through overproduction of ROS. These results suggest that pristimerin has potential therapeutic effects on glioma.
Pristimerin, a natural triterpenoid isolated form Celastrus and Maytenus spp, has been shown to possess a variety of biological and pharmacological effects. Recently, pristimerin has attracted more attention, especially for its potential anticancer activities. The anticancer activities of pristimerin have been illustrated in various cancer cell lines and animal models. It has been found to inhibit in vitro and in vivo proliferation, survival, angiogenesis and metastasis of tumor cells. These activities have been attributed to its modulation of various molecular targets such as cyclins, apoptosis– related proteins, proteasome activity, reactive oxygen species, as well as NF-ΚB, AKT/mTOR and MAPK/ERK pathways. This mini-review discussed the cellular impact and animal studies of pristimerin treatment, with more attention on the various molecular targets of pristimerin.
VEGF INHIBITOR
Pristimerin, a triterpenoid, inhibits tumor angiogenesis by targeting VEGFR2 activation.
Pristimerin is a triterpenoid isolated from Celastrus and Maytenus spp. that has been shown to possess a variety of biological activities, including anti-cancer activity. However, little is known about pristimerin’s effects on tumor angiogenesis. In this study, we examined the function and the mechanism of this compound in tumor angiogenesis using multiple angiogenesis assays. We found that pristimerin significantly reduced both the volume and weight of solid tumors and decreased angiogenesis in a xenograft mouse tumor model in vivo. Pristimerin significantly inhibited the neovascularization of chicken chorioallantoic membrane (CAM) in vivo and abrogated vascular endothelial growth factor (VEGF)-induced microvessel sprouting in an ex vivo rat aortic ring assay. Furthermore, pristimerin inhibited the VEGF-induced proliferation, migration and capillary-like structure formation of human umbilical vascular endothelial cells (HUVECs) in a concentration-dependent manner. Mechanistic studies revealed that pristimerin suppressed the VEGF-induced phosphorylation of VEGF receptor 2 kinase (KDR/Flk-1) and the activity of AKT, ERK1/2, mTOR, and ribosomal protein S6 kinase. Taken together, our results provide evidence for the first time that pristimerin potently suppresses angiogenesis by targeting VEGFR2 activation. These results provide a novel mechanism of action for pristimerin which may be important in the treatment of cancer.
Here, we report that grape seed proanthocyanidins (GSPs) induce apoptosis of NSCLC cells, A549 and H1299, in vitro which is mediated through increased expression of pro-apoptotic protein Bax, decreased expression of anti-apoptotic proteins Bcl2 and Bcl-xl, disruption of mitochondrial membrane potential, and activation of caspases 9, 3 and poly (ADP-ribose) polymerase (PARP). Pre-treatment of A549 and H1299 cells with the caspase-3 inhibitor (z-DEVD-fmk) significantly blocked the GSPs-induced apoptosis of these cells confirmed that GSPs-induced apoptosis is mediated through activation of caspases-3. Treatments of A549 and H1299 cells with GSPs resulted in an increase in G1 arrest. G0/G1 phase of the cell cycle is known to be controlled by cyclin dependent kinases (Cdk), cyclin-dependent kinase inhibitors (Cdki) and cyclins. Our western blot analyses showed that GSPs-induced G1 cell cycle arrest was mediated through the increased expression of Cdki proteins (Cip1/p21 and Kip1/p27), and a simultaneous decrease in the levels of Cdk2, Cdk4, Cdk6 and cyclins. Further, administration of 50, 100 or 200 mg GSPs/kg body weight of mice by oral gavage (5 d/week) markedly inhibited the growth of s.c. A549 and H1299 lung tumor xenografts in athymic nude mice, which was associated with the induction of apoptotic cell death, increased expression of Bax, reduced expression of anti-apoptotic proteins and activation of caspase-3 in tumor xenograft cells. Based on the data obtained in animal study, human equivalent dose of GSPs was calculated, which seems affordable and attainable. Together, these results suggest that GSPs may represent a potential therapeutic agent for the non-small cell lung cancer.
Promoting apoptosis through alterations in Cdki-Cdk-cyclin cascade, and caspase-3 activation via loss of mitochondrial membrane potential
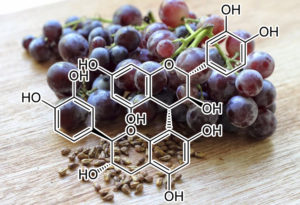
Procyanidin B3
inhibitor of histone acetyltransferase p300
Increasing evidence suggests that AR (androgen receptor) acetylation is critical for prostate cancer cell growth. In the present study, we identified Pro-B3 (procyanidin B3) as a specific HAT (histone acetyltransferase) inhibitor. Pro-B3 selectively inhibited the activity of HATs, but not other epigenetic enzymes. Pro-B3 substantially inhibited the p300-mediated AR acetylation, both in vitro and in vivo. Pro-B3 inhibited both p300-dependent and agonist-induced AR transcription. We demonstrate that the p300-mediated AR acetylation is critical for the hormone responsiveness of AR. Interestingly, B3 treatment efficiently enhanced the antagonist activity of flutamide through suppression of p300 HAT activity, demonstrating that relative p300 activity is critical for the antagonist action. Finally, Pro-B3 treatment inhibited acetylation-dependent prostate cell proliferation and expression of cell-cycle control genes, subsequently increasing cell death, indicating the functional importance of AR acetylation for prostate cancer cell growth.

NF-kB⊥, p65⊥, IL2↓, IkB-α⊥, cyclin B1↑, CDK1↑, cyclin D1↓ p53↑, Bax↑, Bcl-2↓, 1α and cyclin E↓, cdc2↑, cdc2↓, survivin↓, caspase-3↑, COX-2⊥, STAT3, I-κB↓, Tubulin, binding of colchicine to tubulin⊥, bcl-x(L) ⊥, NAG-1↑, JNK↑, ERK↓, Wee1 kinase and p21↑, Bcl-xL↓, Bax↑, caspase-7↑, Fas/APO-1↑, Bcl-2 binding with Beclin 1⊥, Akt phosphorylation↓
Upregulates P53, BAX, Downregulates BCL-2
Pseudolaric acid B (PLAB) is one of the major bioactive components of Pseudolarix kaempferi. It has been reported to exhibit inhibitory effect on cell proliferation in several types of cancer cells. However, there is no report elucidating its effect on glioma cells and organ toxicity in vivo. In the present study, we found that PLAB inhibited growth of U87 glioblastoma cells in a dose-dependent manner with IC(50)~10 μM. Flow cytometry analysis showed that apoptotic cell death mediated by PLAB was accompanied with cell cycle arrest at G2/M phase. Using Western blot, we found that PLAB induced G2/M phase arrest by inhibiting tubulin polymerization in U87 cells. Apoptotic cell death was only partially inhibited by pancaspase inhibitor, z-VAD-fmk, which suggested that PLAB-induced apoptosis in U87 cells is partially caspase-independent. Further mechanistic study demonstrated that PLAB induced caspase-dependent apoptosis via upregulation of p53, increased level of proapoptotic protein Bax, decreased level of antiapoptotic protein Bcl-2, release of cytochrome c from mitochondria, activation of caspase-3 and proteolytic cleavage of poly (ADP-ribose) polymerase (PARP) and caspase-independent apoptosis through apoptosis inducing factor (AIF). Furthermore, in vivo toxicity study demonstrated that PLAB did not induce significant structural and biochemical changes in mouse liver and kidneys at a dose of 25 mg/kg. Therefore, PLAB may become a potential lead compound for future development of antiglioma therapy.

pterostilbene
HIF-1A INHIBITOR
Targeting MTA1/HIF‐1α signaling by pterostilbenein combination with histone deacetylase inhibitor attenuates prostate cancer progression
The metastasis‐associated protein 1(MTA1)/histone deacetylase (HDAC) unit is a cancer progression‐related epigenetic regulator, which is overexpressed in hormone‐refractory and metastatic prostate cancer (PCa). In our previous studies, we found a significantly increased MTA1 expression in a prostate‐specific Pten‐null mouse model. We also demonstrated that stilbenes, namely resveratrol and pterostilbene (Pter), affect MTA1/HDAC signaling, including deacetylation of tumor suppressors p53 and PTEN.In this study, we examined whether inhibition of MTA1/HDAC using combination of Pter and a clinically approved HDACinhibitor, SAHA (suberoylanilide hydroxamic acid, vorinostat), which also downregulates MTA1, could block prostate tumor progression in vivo. We generated and utilized a luciferase reporter in a prostate‐specific Pten‐null mouse model (Pb‐Cre +; Pten f/f; Rosa26 Luc/+) to evaluate the anticancer efficacy of Pter/SAHA combinatorial approach. Our data showed that Pter sensitized tumor cells to SAHA treatment resulting in inhibiting tumor growth and additional decline of tumor progression. These effects were dependent on the reduction of MTA1‐associated proangiogenic factors HIF‐1α, VEGF, and IL‐1β leading to decreased angiogenesis. In addition, treatment of PCa cell lines in vitro with combined Pter and low dose SAHA resulted in more potent inhibition of MTA1/HIF‐1α than by high dose SAHA alone. Our study provides preclinical evidence that Pter/SAHA combination treatment inhibits MTA1/HIF‐1α tumor‐promoting signaling in PCa. The beneficial outcome of combinatorial strategy using a natural agent and an approved drug for higher efficacy and less toxicity supports further development of MTA1‐targeted therapies in PCa.
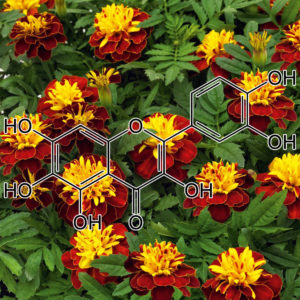
Quercetagetin
Inhibitory effects on cellular senescence
Cellular senescence contributes to tissue and organismal aging, tumor suppression and progress, tissue repair and regeneration, and age-related diseases. Thus, aging intervention might be beneficial for treatment and prevention of diverse age-related diseases. In the present study, we investigated whether four compounds purified from Inula japonica exert inhibitory activity against cellular senescence induced by adriamycin in human umbilical vein endothelial cells (HUVECs). Among them, compound 4 (quercetagetin 3,4′-dimethyl ether) showed inhibitory activity against cellular senescence, which was confirmed by senescence-associated β-galactosidase (SA-β-gal) activity, p53 and p21 protein levels, and intracellular ROS levels. Compound 4 also reduced SA-β-gal activity in HUVECs under replicative senescence. These results suggest that compound 4 represses cellular senescence in HUVECs and might be useful for the development of dietary supplements or cosmetics that alleviate tissue aging or age-related diseases.
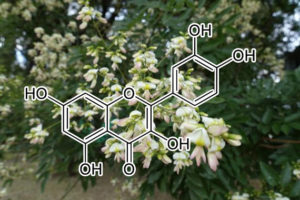
Quercetin
(from sophora japonica aka “Japanese pagoda tree“ and “Chinese scholar tree”)
Direct Binding of Bcl-2 Family Proteins by Quercetin Triggers Its Pro-Apoptotic Activity
Bcl-2 family proteins are important regulators of apoptosis and its antiapoptotic members, which are overexpressed in many types of cancer, are of high prognostic significance, establishing them as attractive therapeutic targets. Quercetin, a natural flavonoid, has drawn much attention because it exerts anticancer effects, while sparing normal cells. A multidisciplinary approach has been employed herein, in an effort to reveal its mode of action including dose–response antiproliferative activity and induced apoptosis effect, biochemical and physicochemical assays, and computational calculations. It may be concluded that, quercetin binds directly to the BH3 domain of Bcl-2 and Bcl-xL proteins, thereby inhibiting their activity and promoting cancer cell apoptosis.
Pleiotropic Effects of Tocotrienols and Quercetin on Cellular Senescence: Introducing the Perspective of Senolytic Effects of Phytochemicals.
The possibility to target cellular senescence with natural bioactive substances open interesting therapeutic perspective in cancer and aging. Engaging senescence response is suggested as a key component for therapeutic intervention in the eradication of cancer. At the same time, delaying senescence or even promote death of accumulating apoptosis-resistant senescent cells is proposed as a strategy to prevent age related diseases. Although these two desired outcome present an intrinsic dichotomy, there are examples of promising natural compounds that appear to satisfy all the requirements to develop senescence– targeted health promoting nutraceuticals. Tocotrienols (T3s) and quercetin (QUE), albeit belonging to different phytochemical classes, display similar and promising effects “in vitro” when tested in normal and cancer cells. Both compounds have been shown to induce senescence and promote apoptosis in a multitude of cancer lines. Conversely, they display senescence delaying activity in primary cells and rejuvenating effects in senescent cells. More recently, QUE has been shown to display senolytic effects in some primary senescent cells, likely as a consequence of its inhibitory effects on specific anti-apoptotic genes (i.e. PI3K and other kinases). Senolytic activity has not been tested for T3s but part of metabolic and apoptotic pathways affected by these compounds in cancer cells overlap with those of QUE. This suggests that the rejuvenating effects of T3s and QUE on pre-senescent and senescent primary cells might be the net results of a senolytic activity on senescent cells and a selective survival of a sub-population of non-senescent cells in the culture. The meaning of this hypothesis in the context of adjuvant therapy of cancer and preventive anti-aging strategies with QUE or T3s is discussed.
AMPK-mediated senolytic and senostatic activity of quercetin surface functionalized Fe3O4 nanoparticles during oxidant-induced senescence in human fibroblasts
Highlights • Quercetin surface functionalized magnetite nanoparticles (MNPQ) were synthesized. • MNPQ eliminated hydrogen peroxide-induced senescent human fibroblasts. • MNPQ limited senescence-associated proinflammatory responses. • Senotherapeutic action of MNPQ was accompanied by increased activity of AMPK. • MNPQ may be useful for senolytic– and senostatic-based anti-aging therapies. Abstract Cellular senescence may contribute to aging and age-related diseases and senolytic drugs that selectively kill senescent cells may delay aging and promote healthspan. More recently, several categories of senolytics have been established, namely HSP90 inhibitors, Bcl-2 family inhibitors and natural compounds such as quercetin and fisetin. However, senolytic and senostatic potential of nanoparticles and surface-modified nanoparticles has never been addressed. In the present study, quercetin surface functionalized Fe3O4 nanoparticles (MNPQ) were synthesized and their senolytic and senostatic activity was evaluated during oxidative stress-induced senescence in human fibroblasts in vitro. MNPQ promoted AMPK activity that was accompanied by non-apoptotic cell death and decreased number of stress-induced senescent cells (senolytic action) and the suppression of senescence-associated proinflammatory response (decreased levels of secreted IL-8 and IFN-β, senostatic action). In summary, we have shown for the first time that MNPQ may be considered as promising candidates for senolytic– and senostatic-based anti-aging therapies.
Senolytic effect of dasatinib and quercetin on human beings Joint Event On World Congress on Tissue Engineering, Stem Cells and Regenerative Medicine & International Conference on Cell and Gene Therapy
Senescent cells are the last generation of cell differentiation. If programmed cell death (apoptosis) is not activated, then it becomes the main source of intoxication for organism. After 36 years their amount is so great, that signs of aging start strongly manifesting. For many years, scientists try to find combination of substances that could kill part of senescent cells. In our experiment we examine the senolytic effect of dasatinib and quercetin on people. Dasatinib is a treatment for oncology diseases, especially widely used when other treatments are no longer working. Quercetin is the vitamin from bioflavonoid group. So as to assess the anti-aging effect of dasatinib and quercetin, we went through clinical trial. For this trial we picked up certain volunteers. Our volunteers were only men participants in spectrum of middle and old ages (36-40 y.o.). We eliminated all female participants, because mutagenous impact of dasatinib on ovicells is not examined properly. As for the male-participants they were recommended to avoid fertilization for the first 3 months after the clinical trial. Generally, 64 men took part in our clinical trial. We classified all these participants into 4 groups, by 16 people for each group. First group of participants had to orally administer 50mg of dasatinib along with 500 mg of quercetin; second group of participants orally administered 50mg of dasatinib and 500 mg of placebo; while third group also took quercetin and placebo, but with different oral dose namely 500mg of quercetin along with 50mg of placebo. The participants of fourth group orally administered two compounds, and both were placebo with a dosage of 500mg and 50mg. These participants of 4 groups must orally administer these compounds once a day after meal for 5 days overall. For the accurate assessment of anti-aging effect of all compounds stair ascending test was done by participants a day prior to the start of trial and 21 days after the end of trial along with medical screening each time. Complete blood count was performed on participants all this time, and also since the start of the test participants’ blood pressure was measured each 10 minutes with 3 overall estimates. By the end of the trial with the help of all the gathered data it was possible to make a solid conclusion, namely among all four group the first group of participants who orally administered 50mg of dasatinib and 500mg of quercetin demonstrated remarkably outstanding improvement of physical endurance as compared to the rest of the groups. The simultaneous oral administration of dasatinib (50mg) and quercetin (500mg) showcased obvious senolytic (anti-aging) effect.
Low-dose quercetin alone improved the healthspan of physiologically aging mice.
In this study, we reported for the first time a geroprotective effect of low-dose quercetin alone that improved the healthspan of aged C57BL/6J male mice. Que-treated mice showed less hair loss, greater athletic endurance, enhanced diastolic function, and less muscle fibrosis, as well as alleviated cellular senescence in multiple tissues. Interestingly, these changes appear to be rarely associated with transcriptional alterations of protein-coding genes but are linked to heterochromatin stabilization and RTE silencing. Que treatment prevented L1 from hyperactivation, thereby inhibiting SASP. In contrast to the reported senolytic effect of high-dose D+Q (Xu et al., 2018), where Que exerts geroprotective effects via the induction of apoptosis of senescent cells, low-dose Que (0.125 mg/kg body weight) alone was sufficient to exert senostatic effects in mice by affecting heterochromatin stability through repression of RTEs activity in this study. In a translational context, low-dose Que monotherapy may be helpful to minimize the dose-dependent side effects compared to high-dose administration and avoid drug interference when used in combination, probably representing a potential therapeutic option for future clinical application (Shoskes et al., 1999). Of note, here we reported the geroprotective effect of Que in male mice and its effect remains to be studied in female mice.
The possible mechanism of low-dose Que in mice may be associated with its function as a heterochromatin stabilizer and its direct inhibitory activity against reverse transcriptase (Ono et al., 1990; Geng et al., 2018). Loss of heterochromatin architecture and genomic instability are two hallmarks of aging (Zhang et al., 2015). In advanced age, the expression of RTEs is often increased, which may in turn contribute to genomic instability and aging-associated cellular defects (De Cecco et al., 2013). Activation of L1 has been implicated in a variety of age-related disorders, including cancer and neurodegenerative diseases. The activation of L1 (and possibly other RTEs in mice) promotes the expression inflammatory factors, a feature of cellular senescence (De Cecco et al., 2019; Simon et al., 2019). Recently, it has been reported that nucleoside reverse-transcriptase inhibitors (NRTIs), such as lamivudine, stavudine, inhibit L1 retrotransposition and thus improve the healthspan and/or lifespan of SIRT6-knockout and physiologically aged mice (De Cecco et al., 2019; Simon et al., 2019). Que has been proven as a potential inhibitor of reverse transcriptase from Rauscher murine leukemia virus (RLV) and human immunodeficiency virus (HIV) by enzyme kinetic analysis, whereas its reverse transcriptase inhibition activity against RTEs in hMSCs and rodents has not been reported (Ono et al., 1990). Our data provide important evidence supporting the role of low-dose Que in safeguarding genomic stability (i.e. inhibition of retrotransposition), which at least in part contributes to its geroprotective activity in rodents.
Quercetin in Idiopathic Pulmonary Fibrosis: Another Brick in the Senolytic Wall
Cellular senescence can be defined as a stable arrest of the cell cycle coupled with stereotyped phenotypic changes (4), including a complex proinflammatory response known as senescence-associated secretory phenotype (SASP) (5). Taking into account recent research from our group and others, it has been postulated that cellular senescence could be a key mechanism in IPF epithelial cells and fibroblasts (2, 6–10). Consequently, senolytic drugs (molecules that induce selective elimination of senescent cells) have been proposed as possible therapies in IPF (11). Various potential senolytic therapies, including dasatinib and quercetin, have been tested in different conditions. The combination of dasatinib and quercetin was shown to induce the clearance of senescent cells and improve lung function in a model of bleomycin lung fibrosis (8).
In this issue of the Journal, Hohmann and colleagues (pp. 28–40) provide additional information on the mechanisms involved in the effect of quercetin alone in lung fibrosis (12). The effectiveness of quercetin alone has already been tested in a model of bleomycin-induced senescence in fibroblasts in vitro, and was found to diminish the proinflammatory expression of SASP (13). After the instillation of bleomycin in a model of lung fibrosis in Wistar rats, the administration of oral quercetin reduced collagen deposition, the antioxidant–oxidant imbalance, and matrix metalloproteinase-7 activity in lung tissues (14). Other studies demonstrated that quercetin reduced the concentration of profibrotic mediators such as IL-13, platelet-derived growth factor β, and TNF-α in BAL fluid obtained from rats with bleomycin-induced pulmonary fibrosis (15). Hohmann and colleagues tested the effectiveness of quercetin in IPF lung fibroblasts and in a model of bleomycin-induced lung fibrosis in aged mice. In agreement with previous studies (7, 8), they confirmed the presence of cellular senescence in IPF fibroblasts. Interestingly, the authors also demonstrated that senescent IPF fibroblasts were highly resistant to FasL- or TRAIL-induced apoptosis and exhibited lower expression of FasL and TNF-related apoptosis-inducing ligand (TRAIL) receptors. Additionally, expression of caveolin-1 (a protein that suppresses cellular senescence and contributes to susceptibility to apoptosis) was decreased, whereas AKT (a modulator of caveolin-1 expression) was activated in IPF fibroblasts. Quercetin reversed all of these effects in IPF senescent cells, suggesting that this senolytic drug could reverse the suppression of death ligand–induced apoptosis, increase FasL and caveolin-1 expression, and inhibit AKT activation. Finally, they demonstrated in aged mice that quercetin mitigated pulmonary fibrosis after 7 days of bleomycin instillation, a time when lung fibrosis is established and inflammatory activators are decreasing. Collectively, these findings provide mechanistic information about the therapeutic effect of quercetin in experimental models of lung fibrosis and its potential use in future research into IPF.
However, the use of senolytics in IPF has some possible limitations that need to be considered (11, 16). First, cellular senescence could be associated with some beneficial processes, such as directing tissue regeneration, limiting the fibrogenic response to acute tissue damage, and promoting the clearance of neoplastic cells through SASP (11). Second, some adverse effects have been attributed to some senolytics. For example, dasatinib can potentially induce pulmonary endothelial damage and increase the risk of pulmonary hypertension (16). Finally, as epithelial cells are a target of senolytics, some authors have hypothesized that the impairment of the epithelial barrier and the potential apoptosis of epithelial cells could induce diffuse alveolar damage (16). To avoid this, the effect of senolytics should be directed to specific sites that are clearly affected by lung fibrosis while avoiding delivery of these drugs to nonaffected areas. Consequently, the complete inhibition of cellular senescence could be avoided, thereby minimizing unwanted effects. A balance between the therapeutic effect and the potential adverse effects of senolytics must be considered in future translational and clinical studies in humans.
In conclusion, Hohmann and colleagues have expanded our knowledge of the mechanisms of quercetin activity as a potential therapeutic senolytic in lung fibrosis. Senolytic therapy is a novel strategy in different diseases that include cellular senescence in their pathogenesis. In recent years, no new therapeutic options have been approved for IPF apart from nintedanib and pirfenidone, so senolytics need to be considered as a novel potential treatment for IPF in future research.
Quercetin-induced apoptosis ameliorates vascular smooth muscle cell senescence through AMP-activated protein kinase signaling pathway
Aging is one of the risk factors for the development of cardiovascular diseases. During the progression of cellular senescence, cells enter a state of irreversible growth arrest and display resistance to apoptosis. As a flavonoid, quercetin induces apoptosis in various cells. Accordingly, we investigated the relationship between quercetin-induced apoptosis and the inhibition of cellular senescence, and determined the mechanism of oxidative stress-induced vascular smooth muscle cell (VSMC) senescence. In cultured VSMCs, hydrogen peroxide (H2O2) dose-dependently induced senescence, which was associated with increased numbers of senescence-associated β-galactosidase-positive cells, decreased expression of SMP30, and activation of p53-p21 and p16 pathways. Along with senescence, expression of the anti-apoptotic protein Bcl-2 was observed to increase and the levels of proteins related to the apoptosis pathway were observed to decrease. Quercetin induced apoptosis through the activation of AMP-activated protein kinase. This action led to the alleviation of oxidative stress-induced VSMC senescence. Furthermore, the inhibition of AMPK activation with compound C and siRNA inhibited apoptosis and aggravated VSMC senescence by reversing p53-p21 and p16 pathways. These results suggest that senescent VSMCs are resistant to apoptosis and quercetin-induced apoptosis attenuated the oxidative stress-induced senescence through activation of AMPK. Therefore, induction of apoptosis by polyphenols such as quercetin may be worthy of attention for its anti-aging effects.
Pleiotropic effects of tocotrienols and Quercetin on cellular senescence: introducing the perspective of Senolytic effects of phytochemicals
The possibility to target cellular senescence with natural bioactive substances open interesting therapeutic perspective in cancer and aging. Engaging senescence response is suggested as a key component for therapeutic intervention in the eradication of cancer. At the same time, delaying senescence or even promote death of accumulating apoptosis-resistant senescent cells is proposed as a strategy to prevent age related diseases. Although these two desired outcome present an intrinsic dichotomy, there are examples of promising natural compounds that appear to satisfy all the requirements to develop senescence– targeted health promoting nutraceuticals. Tocotrienols (T3s) and quercetin (QUE), albeit belonging to different phytochemical classes, display similar and promising effects “in vitro” when tested in normal and cancer cells. Both compounds have been shown to induce senescence and promote apoptosis in a multitude of cancer lines. Conversely, they display senescence delaying activity in primary cells and rejuvenating effects in senescent cells. More recently, QUE has been shown to display senolytic effects in some primary senescent cells, likely as a consequence of its inhibitory effects on specific anti-apoptotic genes (i.e. PI3K and other kinases). Senolytic activity has not been tested for T3s but part of metabolic and apoptotic pathways affected by these compounds in cancer cells overlap with those of QUE. This suggests that the rejuvenating effects of T3s and QUE on pre-senescent and senescent primary cells might be the net results of a senolytic activity on senescent cells and a selective survival of a sub-population of non-senescent cells in the culture. The meaning of this hypothesis in the context of adjuvant therapy of cancer and preventive anti-aging strategies with QUE or T3s is discussed.
Increased renal cellular senescence in murine high-fat diet: effect of the Senolytic drug Quercetin
Obesity and dyslipidemia can be associated with cellular senescence, and may impair kidney function. However, whether senescence contributes to renal dysfunction in these conditions remains unclear. Quercetin is an abundant dietary flavonoid that selectively clears inhibiting PI3K/AKT and p53/p21/serpines and inducing apoptosis. We hypothesized that high-fat-diet-induced obesity causes renal senescence, which would be mitigated by quercetin. C57BL/6J mice fed either standard chow or high-fat diets (HFDs) were treated with quercetin (50 mg/kg) or vehicle 5-days biweekly via oral gavage for 10 weeks. Subsequently, renal function was studied in vivo using magnetic resonance imaging, and renal senescence and histology were evaluated ex vivo. Mice fed with a HFD developed obesity and hypercholesterolemia, whereas renal size remained unchanged. Murine obesity impaired renal function and cortical oxygenation, and induced glomerulomegaly. Renal markers of senescence (eg, expression of p16, p19, and p53) and its secretory phenotype were upregulated in the obese hypercholesterolemic compared to lean mice in renal tubular cells, but attenuated in quercetin-treated murine kidneys, as was renal fibrosis. Quercetin treatment also increased renal cortical oxygenation and decreased plasma creatinine levels in obese mice, whereas body weight and cholesterol levels were unaltered. Therefore, murine obesity and dyslipidemia induce renal tissue senescence and impairs kidney function, which is alleviated by chronic senolytic treatment. These findings implicate senescence in loss of kidney function in murine dyslipidemia and obesity, and support further studies of senolytic therapy in obesity.
Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease
Senescent cells, which can release factors that cause inflammation and dysfunction, the senescence-associated secretory phenotype (SASP), accumulate with ageing and at etiological sites in multiple chronic diseases. Senolytics, including the combination of Dasatinib and Quercetin (D + Q), selectively eliminate senescent cells by transiently disabling pro-survival networks that defend them against their own apoptotic environment. In the first clinical trial of senolytics, D + Q improved physical function in patients with idiopathic pulmonary fibrosis (IPF), a fatal senescence-associated disease, but to date, no peer-reviewed study has directly demonstrated that senolytics decrease senescent cells in humans.
Quercetin-3-O-β-d-glucuronide isolated from Polygonum aviculare inhibits cellular senescence in human primary cells
Cellular senescence is known to contribute to tissue aging, a variety of age-related diseases, tissue regeneration, and cancer. Therefore, aging intervention might be useful for prevention of aging as well as age-related disease. In this study, we investigated compounds from Polygonum aviculare to determine if they inhibited cellular senescence in human primary cells, human dermal fibroblasts (HDFs) and human umbilical vein endothelial cells (HUVECs). Ten compounds from P. aviculare were purified and their inhibitory effects on adriamycin-induced cellular senescence were measured by observing senescence-associated β-galactosidase (SA-β-gal) activity and reactive oxygen species. Among them, compound 9 (quercetin-3-O-β-D-glucuronide) showed inhibitory effects against cellular senescence in HDFs and HUVECs treated with adriamycin. Additionally, compound 9 rescued replicative senescence in HDFs and HUVECs. These data imply that compound 9 represses cellular senescence in human primary cells and might be useful for the development of dietary supplements or cosmetics that ameliorate tissue aging or aging-associated diseases.
Polymeric microsphere-facilitated site-specific delivery of Quercetin prevents senescence of pancreatic islets in vivo and improves transplantation outcomes in mouse Model of Diabetes
Attenuation of senescence progression may be attractive way to preserve the functionality of pancreatic islets (PI) after transplantation. In this study, we developed a model for in vitro induction of premature senescence in rat PI and showed the effectiveness of quercetin (QU) to prevent the senescence.To provide targeted-delivery of QU to the PI after transplantation, we prepared the hybrid clusters (HC) of islet single cells (ISC) and QU-loaded polymeric microspheres (QU; ∼7.55 ng HC−1). Long-term culture of the HC revealed reduced levels of reactive oxygen species and decreased expression of senescence-associated beta galactosidase, Rb, p53, p16, and p21 compared to that of the control islets. Transplantation of HC into subcutaneous space of the immune-deficient mice produced better glycemic control compared to the control islets or the ICC-transplanted mice. SA-β-Gal staining of the in vivo transplanted HC sample showed lower intensity compared to that of the control islets or the islet cell clusters. Thus, in situ delivery of therapeutic agent may be a promising approach to improve therapeutic outcomes in cell therapy.
Inhibition of nitric oxide in LPS-stimulated macrophages of young and senescent mice by δ-tocotrienol and Quercetin
Changes in immune function believed to contribute to a variety of age-related diseases have been associated with increased production of nitric oxide (NO). We have recently reported that proteasome inhibitors (dexamethasone, mevinolin, quercetin, δ-tocotrienol, and riboflavin) can inhibit lipopolysaccharide (LPS)-induced NO production in vitro by RAW 264.7 cells and by thioglycolate-elicited peritoneal macrophages derived from four strains of mice (C57BL/6, BALB/c, LMP7/MECL-1-/- and PPAR-α-/- knockout mice). The present study was carried out in order to further explore the potential effects of diet supplementation with naturally-occurring inhibitors (δ-tocotrienol and quercetin) on LPS-stimulated production of NO, TNF-α, and other pro-inflammatory cytokines involved in the ageing process. Young (4-week-old) and senescent mice (42-week old) were fed control diet with or without quercetin (100 ppm), δ-tocotrienol (100 ppm), or dexamethasone (10 ppm; included as positive control for suppression of inflammation) for 4 weeks. At the end of feeding period, thioglycolate-elicited peritoneal macrophages were collected, stimulated with LPS, LPS plus interferon-β (IFN-β), or LPS plus interferon-γ (IFN-γ), and inflammatory responses assessed as measured by production of NO and TNF-α, mRNA reduction for TNF-α, and iNOS genes, and microarray analysis.
Quercetin Enhances Ligand-induced Apoptosis in senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo
Although cellular senescence may be a protective mechanism in modulating proliferative capacity, fibroblast senescence is now recognized as a key pathogenic mechanism in idiopathic pulmonary fibrosis (IPF). In aged mice, abundance and persistence of apoptosis-resistant senescent fibroblasts play a central role in nonresolving lung fibrosis after bleomycin challenge. Therefore, we investigated whether quercetin can restore the susceptibility of senescent IPF fibroblasts to proapoptotic stimuli and mitigate bleomycin-induced pulmonary fibrosis in aged mice. Unlike senescent normal lung fibroblasts, IPF lung fibroblasts from patients with stable and rapidly progressing disease were highly resistant to Fas ligand (FasL)-induced and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Senescent IPF fibroblasts exhibited decreased expression of FasL and TRAIL receptors and caveolin-1, as well as increased AKT activation, compared with senescent normal lung fibroblasts. Although quercetin alone was not proapoptotic, it abolished the resistance to FasL- or TRAIL-induced apoptosis in IPF fibroblasts. Mechanistically, quercetin upregulated FasL receptor and caveolin-1 expression and modulated AKT activation. In vivo quercetin reversed bleomycin-induced pulmonary fibrosis and attenuated lethality, weight loss, and the expression of pulmonary senescence markers p21 and p19-ARF and senescence-associated secretory phenotype in aged mice. Collectively, these data indicate that quercetin reverses the resistance to death ligand–induced apoptosis by promoting FasL receptor and caveolin-1 expression and inhibiting AKT activation, thus mitigating the progression of established pulmonary fibrosis in aged mice. Therefore, quercetin may be a viable therapeutic option for IPF and other age-related diseases that progress with the accumulation of senescent fibroblasts.
Restoring effects of natural anti-oxidant Quercetin on cellular senescent human dermal fibroblasts
The oxidative damage initiated by reactive oxygen species (ROS) is a major contributor to the functional decline and disability that characterizes aging. The anti-oxidant flavonoid, quercetin, is a plant polyphenol that may be beneficial for retarding the aging process. We examined the restoring properties of quercetin on human dermal fibroblasts (HDFs). Quercetin directly reduced either intracellular or extracellular ROS levels in aged HDFs. To find the aging-related target genes by quercetin, microarray analysis was performed and two up-regulated genes LPL and KCNE2 were identified. Silencing LPL increased the expression levels of senescence proteins such as p16INK4A and p53 and silencing KCNE2reversed gene expressions of EGR1 and p-ERK in quercetin-treated aged HDFs. Silencing of LPL and KCNE2 decreased the expression levels of anti-oxidant enzymes such as superoxide dismutase and catalase. Also, the mitochondrial dysfunction in aged HDFs was ameliorated by quercetin treatment. Taken together, these results suggest that quercetin has restoring effect on the cellular senescence by down-regulation of senescence activities and up-regulation of the gene expressions of anti-oxidant enzymes in aged HDFs.
Protective effect of Quercetin-quinone derivative in senescent human fibroblasts
Gradual accumulation of oxidatively damaged cell structures along with impairment of redox homeostasis has been suggested as a key factor in ageing and cellular senescence. Anti-ageing effects were reported for a number of phenol- and quinone-type compounds. By using a model of senescent human fibroblasts (strain VH-10), potential beneficial effects of semisynthetic compound, 4-O-(2-chloro-1,4-naphtoquinone-3-yloxy) quercetin (CHNQ) were investigated. CHNQ significantly suppressed senescence markers, SAβ-galactosidase activity and lipofuscin-related autofluorescence. Moreover, CHNQ, more efficiently than its precursors, restored mitochondrial membrane potential linked probably to observed decrease in ROS levels. In addition, CHNQ-treated cells showed increased LC3 turnover pointing to improvement of autophagy flux. By contrast, the compounds tested restored neither the gradual decline in proliferation, nor the cell cycle arrest at G2/M phase. Yet, modulation of p21 and p53 proteins dependently on the persistence time of senescent cells in culture was found. In conclusion, the result of our study point to protective effect of CHNQ on functionalities of senescent fibroblasts, without influencing their proliferative lifespan [VEGA 2/0041/17,2/0029/16,APVV-15–0308, ITMS 26240220040].
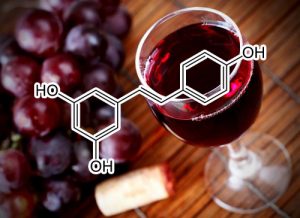
Resveratrol
Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells
Signal transducer and activator of transcription 3 (STAT3) is persistently activated in squamous cell carcinoma of the head and neck (SCCHN) and can cause uncontrolled cellular proliferation and division.We found that HN3 and FaDu cells expressed strongly phosphorylated STAT3 on both tyrosine 705 and serine 727 residues as compared to other SCCHN cells. The phosphorylation was completely suppressed by resveratrol in FaDu cells, but not substantially in HN3 cells. STAT3 suppression was mediated through the inhibition of activation of upstream JAK2, but not of JAK1 and Src kinases. Treatment with the protein tyrosine phosphatase (PTP) inhibitor pervanadate reversed the resveratrol-induced down-regulation of STAT3, thereby indicating a critical role for a PTP. We also found that resveratrol induced the expression of the SOCS-1 protein and mRNA. Further, deletion of SOCS-1 gene by siRNA suppressed the induction of SOCS-1, and reversed the inhibition of STAT3 activation. Resveratrol down-regulated various STAT3-regulated gene products, inhibited proliferation, invasion, as well as induced the cell accumulation in the sub-G1 phase and caused apoptosis. Beside, this phytoalexin also exhibited the enhancement of apoptosis when combined with ionizing radiation treatment.
Conclusion
Our results suggest that resveratrol blocks STAT3signaling pathwaythrough induction of SOCS-1, thus attenuating STAT3 phosphorylation and proliferation in SCCHN cells.
Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: effects on the inhibition of STAT3 phosphorylation.
Resveratrol, a polyphenolic phytoaxelin present in red wine, has been suggested to protect against atherosclerosis and cardiovascular disease because of its antioxidant effects. Intercellular adhesion molecule (ICAM-1), induced by cytokines, has been hypothesized to play a role in the early events during atherosclerosis. In this study we tested the effects of resveratrol upon both IL-6-induced ICAM-1 gene expression and its underlying signaling pathways in endothelial cells (ECs). Resveratrol was found to inhibit both TNFalpha- and IL-6-induced ICAM-1 gene expression at the promoter, transcriptional and protein levels. Resveratrol also abrogates the tyr705 phosphorylation of STAT3 in IL-6-treated ECs, in a dose- and time-dependent manner. Although quercetin had similar effects, resveratrol showed higher inhibitory properties following 2-4 h pretreatments. Resveratrol has been shown to induce the activity of endothelial nitric oxide synthase (eNOS) and increase NO production. Consistent with this, the treatment of ECs with a NO donor (SNAP) reduces IL-6-induced STAT3 phosphorylation. Conversely, exposure of ECs to a NOS inhibitor reversed the effects of resveratrol upon IL-6-induced STAT3 phosphorylation. Furthermore, ECs transfected with constitutively active Rac1 (RacV12) showed increases in ICAM-1 promoter activity, intracellular reactive oxygen species (ROS) levels and STAT3 phosphorylation, and these increases were attenuated by resveratrol treatment. In summary, we demonstrate for the first time that resveratrol inhibits IL-6-induced ICAM-1 gene expression, in part, by interfering with Rac-mediated pathways via the attenuation of STAT3 phosphorylation. This study therefore provides important new insights that may contribute to the proposed beneficial effects of resveratrol in endothelial responses to cytokines during inflammation.
Resveratrol addiction: To die or not to die
Cell killing by this stilbene may be mediated through any of numerous mechanisms that involve activation of mitochondria and of death caspases; upregulation of cyclin‐dependent kinase inhibitors, tumor suppressor gene products, or death‐inducing cytokines and cytokine receptors; or downregulation of cell survival proteins (survivin, cFLIP, cIAPs, X‐linked inhibitor of apoptosis protein (XIAP), bcl‐2, bcl‐XL) or inhibition of cell survival kinases (e.g., mitogen‐activiated protein kinases (MAPKs), AKT/phosphoinositide 3‐kinase (PI3K), PKC, EGFR kinase) and survival transcription factors (nuclear factor‐kappaB (NF‐κB), activating protein 1 (AP‐1), HIF‐1α, signal transducer and activator of transcription (STAT3)).
Enhanced inhibition of adipogenesis and induction of apoptosis in 3T3-L1 adipocytes
Certain flavonoids have been shown to have specific effects on biochemical and metabolic functions of adipocytes. In this study, we investigated the effects of combinations of resveratrol and quercetin on adipogenesis and apoptosis in 3T3-L1 cells. In maturing preadipocytes resveratrol and quercetin at 25 μM individually suppressed intracellular lipid accumulation by 9.4 ± 3.9% (p < 0.01) and 15.9 ± 2.5%, respectively, (p < 0.001). The combination of resveratrol and quercetin at the same dose, however, decreased lipid accumulation by 68.6 ± 0.7% (p < 0.001). In addition, combinations of resveratrol and quercetin at 25 μM significantly decreased the expression of peroxisome proliferators-activated receptor gamma (PPARγ) and CCAAT/enhancer-binding protein (C/EBP)α, both of which act as key transcription factors. In mature adipocytes resveratrol and quercetin at 100 μM individually decreased viability by 18.1 ± 0.6% (p < 0.001) and 15.8 ± 1% (p < 0.001) and increased apoptosis (100 μM) by 120.5 ± 8.3% (p < 0.001) and 85.3 ± 10% (p < 0.001) at 48 h, respectively. Combinations of resveratrol and quercetin further decreased viability (73.5 ± 0.9%, p < 0.001) and increased apoptosis (310.3 ± 9.6%, p < 0.001) more than single compounds alone. The combination of resveratrol and quercetin at 100 μM increased release of cytochrome c from mitochondria to cytosol and decreased ERK 1/2 phosphorylation. Taken together, our data indicate that combinations of resveratrol and quercetin can exert potential anti-obesity effects by inhibiting differentiation of preadipocytes and inducing apoptosis of mature adipocytes.

Rottlerin
(from Mallotus philippinensis aka “Red Kamala Tree”)
BCL Inhibitor
Rottlerin stimulates apoptosis both in PaCa cells in vitro and in vivo through inhibition of Bcl-xL. Thus, rotterlin represents a promising agent for pancreatic cancer treatment.
Downregulates BCL-2, BCL-XL, XIAP, IAP1
Prolonged exposure of breast CSCs to Rottlerin caused apoptosis (associated with down‐regulation of anti‐apoptotic Bcl‐2, Bcl‐X(L), XIAP and cIAP‐1), and autophagy‐type behaviour such as suppression of phosphorylated Akt and mTOR and up‐regulation of phosphorylated AMPK. A further study has shown that Rottlerin induces autophagy viaextensive cytoplasmic vacuolization in breast CSCs. Rottlerin‐induced autophagy can lead to apoptosis in breast CSCs, while Rottlerin treatment increases expression of LC3, Beclin‐1 and ATG12 that accumulate during autophagy 36. Although, autophagy inhibitors suppress formation of cytoplasmic vacuolization and autophagy in CSCs, the potency of Rottlerin to induce autophagy and apoptosis might be based on its capability to activate several pathways such as AMPK and proteasome inhibition 36.
inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP
Treatment with rottlerin significantly decreased Cdc2 activity through the downregulation of cyclin A, cyclin B, and Cdc2 proteins, whereas the sensitizing effect of rottlerin on TRAIL-induced apoptosis was independent of PKCδ activity. Furthermore, treatment with rottlerin downregulated the protein levels of survivin and X-chromosome-linked IAP (XIAP), two major caspase inhibitors. Forced expression of Cdc2 together with cyclin B attenuated rottlerin-potentiated TRAIL-induced apoptosis by over-riding the rottlerin-mediated downregulation of survivin and XIAP protein levels. Taken together, inhibition of Cdc2 activity and the subsequent downregulation of survivin and XIAP by subtoxic doses of rottlerincontribute to amplification of caspase cascades, thereby overcoming resistance of glioma cells to TRAIL-mediated apoptosis. Since rottlerin can sensitize Bcl-2– or Bcl-xL-overexpressing glioma cells but not human astrocytes to TRAIL-induced apoptosis, this combined treatment may offer an attractive strategy for safely treating resistant gliomas.
CYCLIN D1 INHIBITOR
The growth arrest evoked by Rottlerin was not mediated by cell-cycle inhibitors p21 and p27 but was accompanied by a dramatic fall in the cyclin-D1 protein, the levels of which were not altered by the pan-PKC inhibitor GF 109203X, thus excluding a PKC-mediated mechanism in the Rottlerin effect. The parallel drop in cyclin-D1 mRNA suggested a down-regulation of the gene caused by the inhibition of nuclear factor-kappa B (NFκB), which occurs via a PKC-, Akt-, ERK- and mitochondrial uncoupling-independent mechanism. We provide preliminary evidence that the interference on the NFκB activation process likely occurs at the level of calcium/calmodulin-dependent protein kinase II (CaMKII), a known Rottlerin target. Indeed the drug prevented calcium-induced CaMKII autophosphorylation which, in turn, led to decreased NFκB activation.
HDAC INHIBITOR
The polyphenolic alcohol resveratrol has demonstrated promising activities for the prevention and treatment of cancer. Different modes of action have been described for resveratrol including the activation of sirtuins, which represent the class III histone deacetylases (HDACs). However, little is known about the activity of resveratrol on the classical HDACs of class I, II and IV, although these classes are involved in cancer development or progression and inhibitors of HDACs (HDACi) are currently under investigation as promising novel anticancer drugs. We could show by in silico docking studies that resveratrol has the chemical structure to inhibit the activity of different human HDAC enzymes. In vitro analyses of overall HDAC inhibition and a detailed HDAC profiling showed that resveratrol inhibited all eleven human HDACs of class I, II and IV in a dose-dependent manner. Transferring this molecular mechanism into cancer therapy strategies, resveratrol treatment was analyzed on solid tumor cell lines. Despite the fact that hepatocellular carcinoma (HCC) is known to be particularly resistant against conventional chemotherapeutics, treatment of HCC with established HDACi already has shown promising results. Testing of resveratrol on hepatoma cell lines HepG2, Hep3B and HuH7 revealed a dose-dependent antiproliferative effect on all cell lines. Interestingly, only for HepG2 cells a specific inhibition of HDACs and in turn a histone hyperacetylation caused by resveratrol was detected. Additional testing of human blood samples demonstrated a HDACi activity by resveratrol ex vivo. Concluding toxicity studies showed that primary human hepatocytes tolerated resveratrol, whereas in vivo chicken embryotoxicity assays demonstrated severe toxicity at high concentrations. Taken together, this novel pan- HDACi activity opens up a new perspective of resveratrol for cancer therapy alone or in combination with other chemotherapeutics. Moreover, resveratrol may serve as a lead structure for chemical optimization of bioavailability, pharmacology or HDAC inhibition.
Rutin
Saussurea involucrata (Kar. et Kir.), known as the snow lotus, grows in the Tian Shan and A’er Tai areas of China. It has recently been reported that the ethyl acetate extract of S. involucrata (SI-2) can inhibit proliferation and induce apoptosis in PC-3 human prostate cancer cells. This study investigated the protective effect of ethyl acetate extract of S. involucrata (SI-2) or rutin, a flavonoid extracted from ethyl acetate extract of S. involucrata (SI-2), on D-galactose- (D-gal-) induced brain injury in mice. Administering SI-2 or rutin (30 mg/kg/d and 30 mg/kg/d) for 6 weeks, concomitant with D-gal injection, significantly increased superoxide dismutase and glutathione peroxidase activities and decreased the MDA level in plasma. Furthermore, the result showed that the percentages of cleaved caspase-3 and PARP in the D-gal-treated mice were much higher than those in the control. Pretreatment using SI-2 or rutin decreased the expression of cyclooxygenase-2 via downregulation of NF-kappaB, resulting in a decrease in lipid peroxidation. Furthermore, our results also showed that oral administration of rutin to these mice significantly improved behavioral performance in a step-through passive avoidance task and these results suggest that SI-2 or rutin exerts potent antiaging effects on D-gal in mice via antioxidative mechanisms.
Inhibition of premature senescence with rutin
Atherosclerosis is an age-associated disease; however, diabetic atherosclerosis has higher severity beyond age range for accumulative premature senescent cells in diabetes. Recent findings suggest that rutin, a flavonoid, has potential benefits for diabetic individuals. This study was designed to evaluate the effects of rutin on premature senescence and atherosclerosis. Apolipoprotein E knockout miceexhibiting insulin resistance after 6 weeks of high-fat diet were administered with a low dose of streptozotocin (STZ) to induce diabetes. After 8 weeks of STZ administration, rutin (40 mg/kg/d) was supplemented by gavage for the last 6 weeks. We evaluated the prosperity of the plaque and diabetes using serial echocardiography, histopathologic and metabolite analysis. Premature senescence induced by hydrogen peroxide in primary vascular smooth muscle cells (VSMCs) was used to analyze the underlying mechanism. Mice with diabetes showed more severe plaque burden on aortic arteries and less smooth muscle cells but larger senescent cell ratio in plaque compared with mice with control diets. Rutin significantly improves glucose and lipid metabolic disturbance in diabetes. Moreover, rutin decreased the atherosclerotic burden and senescent cell number and increased the VSMC ratio in aortic root plaque. In vitro, we demonstrated that rutin ameliorated premature senescence induced by oxidative stress, and the protective function may be mediated by inhibiting oxidative stress and protecting telomere. Rutin administration attenuates atherosclerosis burden and stabilizes plaque by improving metabolic disturbance and alleviating premature senescence of VSMCs. Inhibition of VSMCs premature senescence with rutin may be an effective therapy for diabetic atherosclerosis.

Salvia miltiorrhizae
TLR4 INHIBITOR
Salvia miltiorrhizae is able to reduce the levels of plasma endotoxin and inhibit effectively the expressions of TLR4 protein
downregulates CDC2
Tanshinone IIA inhibits gastric carcinoma AGS cells through increasing p-p38, p-JNK and p53 but reducing p-ERK, CDC2 and cyclin B1 expression.
Tanshinone IIA (Tan-IIA) is extracted from Danshen (Salviae miltiorrhizae radix). It possesses antitumor activity against a variety of human cancer cells and its induction of apoptosis and inhibition of proliferation of gastric cancer cells are well-documented. However, the molecular mechanisms by which Tan-IIA inhibits gastric cancer have not been well-elucidated. In the present study, we evaluated the cytotoxicity of Tan-IIA against human gastric cancer AGS cells by the (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) MTT assay. The protein expression of tumor necrosis factor-alpha (TNF-α), FAS, p53, p21, cyclin A, cyclin B1, extracellular-related kinase (ERK), phospho extracellular-related kinase (p-ERK), p38, p-p38, Jun-amino-terminal kinase (JNK), phospho Jun-amino-terminal kinase (p-JNK) and β-actin in AGS cells were measured by western blotting. The cell-cycle distribution was analyzed by flow cytometry. The results showed that Tan-IIA inhibited AGS cells with time- and dose-dependent manners. AGS cells treated with Tan-IIA up-regulated the protein expression of TNFα, FAS, p-p38, p-JNK, p53, p21, caspase-3 and caspase-8 but reduced that of p-ERK, CDC2, cyclin A, and cyclin B1. The results also showed that Tan-IIA dose dependently induced G2/M phase arrest. These findings demonstrate that Tan-IIA can inhibit AGS human gastric cancer cells; one of the molecular mechanisms may be through increasing the protein expression of p-p38 and p-JNK, but decreasing that of p-ERK to induce the activation of p53, followed by increasing the protein expression of p21 to down-regulate CDC2 and cyclin B1 expression which then induces G2/M phase arrest. Another route may be through increasing the protein expression of TNF-α, FAS, caspase-8 and caspase-3 to induce apoptosis.

Sansalvamide A
(a cyclopeptide from marine fungi Fusarium)
HSP90 INHIBITOR
Described are the syntheses of 3 Sansalvamide A derivatives that contain biotinylated tags at individual positions around the macrocycle. The tagged derivatives indicated in protein pull-down assays that they bind to Hsp90 at the same binding site (N-middle domain) as the San A-amide peptide. Further, these compounds inhibit binding between Hsp90 and multiple C-terminal client proteins. This interaction is unique to the San A analogs indicating they can be tuned for selectivity against Hsp90client/co-chaperone proteins.
Scutellarin
regulates Bcl-2 and Bax expression
The inactivation of Bcl-2 and the activation of Bax commit cancer cells to undergo apoptosis (10). Thus, the effect of Scutellarin on the expression levels of Bcl-2 and Bax in HCT-116 cells were investigated. HCT-116 cells were exposed to 10, 30 and 100 µM Scutellarin for 48 h, and the expression of Bcl-2 protein was significantly decreased when compared with untreated HCT-116 cells (P=0.3036, P=0.0890 and P=0.0093, respectively; Fig. 3A). Consistently, the expression of Bax protein in HCT-116 cells was increased significantly in the presence of 10 (P=0.011), 30 (P=0.0003) and 100 µM (P=0.0002) Scutellarin when compared with the control. The changes in the levels of Bcl-2 and Bax protein expression in HCT-116 cells were associated with the concentration of Scutellarin used. Additionally, the active form of caspase-3 protein was also increased in HCT-116 treated with Scutellarin (Fig. 3B). These results suggest that Scutellarin induces apoptosis of HCT-116 cells via regulating Bcl-2/Bax expression and activating caspase-3.
Silybin / SILIBININ
(the major active constituent of silymarin, a standardized extract of Silybum marianum aka “Milk Thistle Seeds”)
Silibinin inhibits constitutive activation of Stat3, and causes caspase activation and apoptotic death of human prostate carcinoma DU145 cells.
Transcription factor signal transducer and activator of transcription (Stat)-3 is activated constitutively in prostate cancer (PCA) suggesting that its disruption could be an effective approach to control this malignancy. Here we assessed whether silibinin, a flavanone from Silybum marianum with proven anticancer efficacy in various cancer models, inhibits Stat3 activation in DU145 cells, and if it does, what is the biological fate of the cells? At 50 muM or higher concentrations for 24 or 48 h, silibinin concentration dependently reduced constitutive Stat3 phosphorylation at Tyr705 and Ser727 residues under both serum and serum-starved conditions. Constitutively active Stat3-DNA binding was also inhibited concentration dependently by silibinin; however, apoptotic death together with caspase and poly(ADP-ribose) polymerase (PARP) cleavage was observed by silibinin only under serum-starved conditions suggesting that additional survival pathways are active under serum conditions. In other studies, cells were treated with various specific pharmacological inhibitors where phosphorylation of Stat3 was not reduced by epidermal growth factor receptor and Mitogen activated protein/extracellular signal regulate kinase kinase (MEK1/2) inhibitors, suggesting lack of significant roles of these in Stat3 activation in DU145 cells. Janus kinase (JAK)-1 and JAK2 inhibitors strongly reduced Stat3 phosphorylation but did not result in apoptotic cell death. Interestingly, JAK1 inhibitor only in combination with silibinin resulted in a complete reduction in Stat3 phosphorylation at Tyr705, activated caspase-9 and caspase-3, and caused strong PARP cleavage and apoptotic death of DU145 cells. Given a critical role of Stat3 activation in PCA, our results showed that silibinin inhibits constitutively active Stat3 and induces apoptosis in DU145 cells, and thus might have potential significance in therapeutic intervention of this deadly malignancy.
silibinin down-regulatES survivin.
PURPOSE: Chemoprevention is an upcoming approach to control bladder cancer, which is one of the commonly diagnosed malignancies showing recurrence rate of 70% or even higher. Recently, we observed the in vitro efficacy of silibinin, a flavanolignan, in human bladder transitional cell papilloma RT4 cells. Here, we investigated the antitumor efficacy and associated mechanisms of silibinin in RT4 tumor xenograft.
EXPERIMENTAL DESIGN: RT4 tumor xenograft was implanted s.c. in athymic nude mice, and then animals were oral gavaged with silibinin at 100 and 200 mg/kg doses, 5 days/week for 12 weeks. Tumor growth, body weight, and diet consumption were recorded, and tumors were analyzed for proliferation, apoptosis, and angiogenesis biomarkers and molecular alterations by immunohistochemistry, immunoblot analysis, and ELISA. p53 small interfering RNA was used in cell culture to examine the role of p53 in survivin expression.
RESULTS: Silibinin feeding inhibited tumor xenograft growth without any gross signs of toxicity. Silibinin decreased tumor volume by 51% to 58% (P <or= 0.01) and tumor weight by 44% to 49% (P < 0.05). Silibinin moderately (P < 0.001) decreased cell proliferation and microvessel density and strongly (P < 0.001) increased apoptosis in tumors. Silibinin robustly decreased survivin protein expression and its nuclear localization, as well as tumor-secreted level in mouse plasma, but increased p53 and cleaved caspase-3 levels in tumors. Silibinin-caused decrease in survivin was independent of p53.
CONCLUSION: These findings identified in vivo antitumor efficacy of silibinin against human bladder tumor cells involving down-regulation of survivin and an increase in p53 expression together with enhanced apoptosis.
inhibition of proteins of MAPK family and down-regulation of NF-kβ expression
Silymarin is a flavonolignan compound which is abundantly found in the extract of milk thistle plant. In particular, silymarin is a mixture of mainly three flavonolignans, silybin (or silibinin), silydianin, and silychristin. Silibinin is considered the most active component of silymarin, however there are no significant difference between silymarin and silibinin in terms of biological activities [82]. Several in vitro and in vivo animal studies have shown skin anti-aging and photoprotective efficacy of silymarin against UVRinduced inflammation, oxidative stress, and DNA damage. For example, it was observed that the topical application of silymarin to SKH-1 hairless mice provided protection against different stages of photocarcinogenesis, possibly via its strong antioxidant properties [83]. Further results showed that the efficacy of silibinin against photocarcinogenesis, involves the inhibition of DNA synthesis, cell proliferation, and cell cycle progression and an induction of apoptosis [84]. Additional in vivo studies clearly show that the cytoprotective activities of silymarin and/or silibinin against inflammation associated skin diseases also involve inhibition of proteins of MAPK family and down-regulation of NF-kβ expression [85]. Although experimental findings may be clinically relevant, there is a need to translate these results into clinical studies.
Decreases CDK2, CDK4, cyclin E, and cyclin D1; upregulates p27 and p21; attenuates ERK and Akt; suppresses iNOS, COX, HIF-1α, and VEGF; upregulates caspase-3, -8, and -9; decreases β-catenin and Gsk-β levels; decreases c-Myc; suppresses MMP-2 and AP-1; suppresses IL-1; downregulates Bcl-2 and upregulates Bax; targets β-catenin and IGF-1
Silibinin is a flavonone that has been isolated from milk thistle 45. The anticolorectal effect of this compound was extensively studied in various colon cancer cell lines such as HT-29, HCT-116, SW480, and LoVo. Silibinin has been shown to have the potential to inhibit HT-29 cells through different pathways 46. In the presence of silibinin, cell cycle arrest occurred in G2/M. In mechanistic studies related to cell cycle progression, silibinin was associated with decreasing levels of CDK2, CDK4, cyclin E, and cyclin D1 and with the upregulation of p27 and p21 47. Although the antiproliferation and apoptotic effects of silibinin on HT-29 cells likely depend on attenuation of ERK and Akt, suppression of iNOS, COX, hypoxia-inducible factor-1 alpha (HIF-1α), and vascular endothelial growth factor (VEGF) also leads to an antiangiogenesis effect 48.
The inhibitory effect of silibinin on SW480 was reported to occur through four mechanisms: i) cell death and apoptosis via upregulation of caspase-3, -8, and -9 49; ii) the Wnt-β-catenin pathway, whereby silibinin decreases β-catenin and Gsk-β levels; iii) a decrease in angiogenesis regulators such as VEGF and iNOS; and iv) targeting of signaling molecules involved in proliferation and survival such as cyclin D and c-Myc 50. Although silibinin has the potential to inhibit SW480 through apoptosis, growth inhibition of HCT-116 cells in the presence of this compound was independent of apoptosis and more related to suppression of p27, p21, cyclin B1, cyclin D1, and CDK2 46. Another study conducted in 2012 demonstrated that silibinin also had inhibitory effect on LoVo colon cancer cells and that this effect occurred through the suppression of matrix metalloproteinase (MMP)-2 and AP-1 binding activity 51.
In vivo studies of Wistar rats and A/J mice induced by AOM also confirmed the anti-colorectal activity of silibinin. These extensive studies were performed to the level of gene expression and indicated that silibinin might be used as a chemopreventive compound in the battle against CRC in Wistar rats through two mechanisms: i) suppression of inflammatory receptors such as interleukin-1 (IL-1) and ii) induction of apoptosis via the downregulation of Bcl-2 and the upregulation of Bax 52. The chemopreventive mechanism of silibinin was also under investigation in A/J mice. Silibinin targeted β-catenin and IGF-1 in the Wnt and PI3K/Akt pathways, respectively 53.
ACTIVATES BNIP3
BNIP3 is a mitochondrial BH3-only protein that contributes to cell death through activation of the mitochondrial pathway of apoptosis (11). Although it belongs to the Bcl-2 family, its pro-cell death activity is distinct from other family members. BNIP3 is also known to induce mitochondrial autophagy (9). In the present study, our results showed that silencing of BNIP3 significantly reduced the silibinin-enhanced autophagy-related LC3-II/LC3-I ratio, implying an essential role of BNIP3 in silibinin-induced autophagic cell death in MCF7 cells. Notably, BNIP3 siRNA sustained ΔΨm and ATP levels during autophagy, and prevented ROS production in the silibinin-treated MCF7 cells. Similar to what we observed, Ghavami et al showed that overexpression of ∆TM-BNIP3, a dominant-negative BNIP3 mutant which prevents wild-type (wt) BNIP3 from targeting the mitochondria and antagonizes wt BNIP3-induced effects, not only reversed cell death and autophagy, but also reduced ROS production and mitochondrial damage in S100A8/A9-treated cells including MCF7 (39).
In conclusion, the present study shed new light on the mechanisms involved in silibinin-triggered cell death. The present study suggests that silibinin-induced autophagic cell death involves an increase in ROS production, subsequently followed by mitochondrial dysfunction and ATP depletion. Finally, we suggest that BNIP3 is the critical factor that mediates the ROS-dependent mitochondrial death pathway (Fig. 5).
sinapinic acid (Hydnophytum formicarum Jack)
The results in this report demonstrated that ethanolic crude extract and phenolic-rich extract from H. formicarum Jack. rhizome inhibited HDAC activity both in vitro and in the cells. Sinapinic acid was identified as the major component of phenolic extract, which may underpin, at least in part, its HDAC inhibitory activity. The growth inhibitory effect on a cervical cancer cell line (HeLa cells) of ethanolic crude extract, phenolic extract and sinapinic acid is in accordance with their capability to induce cancerous cell apoptosis. Our findings may validate the use of H. formicarum Jack. rhizome extracts as an alternative medicine for cancer treatment. Further investigation, with details about chemical structure modification of sinapinic acid, HDAC inhibitory activity, anticancer activity and combination with other anticancer drugs, is of interest.

Solidago virgaurea
Senolytic activity of goldenrod extract
In 2018, Lämmermann et al. identified an extract from the plantSolidago virgaurea subsp. alpestris, 1201, that can block the negative effects of senescence in human skin fibroblasts, including the SASP in vitro (Lämmermann et al., 2018). 1201 was not only able to delay the acquisition of a senescent phenotype, but was also able to reduce the cell numbers of SCs by 30%, with the senolytic effect of 1201 being mediated by the induction of apoptosis. Interestingly, when 1201 was added to the medium during the differentiation process of the epi- dermal layer, it resulted in an increase in the epidermal thickness compared to untreated controls. Thus, the natural plant extract may represent a promising possibility to block age-related loss of tissue functionality.

Sulforaphane
HDAC INHIBITOR
Consistently, in vivo administration of sulforaphane inhibited HDAC activity in mouse colonic mucosa after six hours from the oral treatment with concomitant increase of acetylated H3 and H4 histone. Changes in histone acetylation status were also observed after long-term (10 weeks) administration of sulforaphane diet that resulted in augmented acetylated histones and p21 expression in the ileum, colon, prostate, and PBMC cells. Dietary sulforaphane was also able to suppress polyp formation in Apcmin mice [203]. In addition, changes in the histone modifications of the hTERT promoter and DNA demethylation of hTERT exon 1 were observed in human breast cancer cells in response to sulforaphane [204]. Acting as HDAC inhibitor, sulforaphane may be useful in the treatment of many types of cancer in which HDAC activity and hypoacetylation contribute to malignant progression.
TLR4 SUPPRESSOR
Sulforaphane Suppresses Oligomerization of TLR4in a Thiol-Dependent Manner
TLRs are pattern recognition receptors that detect invading microorganisms and nonmicrobial endogenous molecules to trigger immune and inflammatory responses during host defense and tissue repair. TLR activity is closely linked to the risk of many inflammatory diseases and immune disorders. Therefore, TLR signaling pathways can provide efficient therapeutic targets for chronic diseases. Sulforaphane (SFN), anisothiocyanate, has been well known forits anti-inflammatory activities. In this study, weinvestigated the modulation of TLR activity by SFN and the underlying mechanism. SFN suppressed ligand-induced and ligand-independent TLR4 activation because it prevented IL-1R–associated kinase-1 degradation, activation of NF-kB and IFN regulatory factor 3, and cyclooxygenase-2 expression induced by LPS or overexpression of TLR4. Receptor oligomerization, which is one of the initial and critical events of TLR4 activation, was suppressed by SFN, resulting in the downregulation of NF-kB activation. SFN formed adducts with cysteine residues in the extracellular domain of TLR4 as confirmed by liquid chromatography-tandem mass spectrometry analysis and the inhibitory effects of SFN on oligomerization and NF-kB activation were reversed by thiol donors (DTT and N-acetyl-L-cysteine). These suggest that the reactivity of SFN to sulfhydryl moiety contributes to its inhibitory activities. Blockade of TLR4 signaling by SFN resulted in the reduced production of inflammatory cytokines and the decreased dermal inflammation and edema in vivo in experimental inflammatory animal models. Collectively, our results demonstrated that SFN downregulated TLR4 signaling through the suppression of oligomerization process in a thiol-dependent manner. These present a novel mechanism for beneficial effects of SFN and a novel anti-inflammatory target in TLR4 signaling.
Sulforaphane and myricetin act synergistically to induce apoptosis in 3T3‑L1 adipocytes
The aim of the present study was to investigate whether sulforaphane (SFN) and myricetin (Myr) synergistically induce apoptosis in adipocytes. The viability of mature 3T3‑L1 adipocytes treated with 40 µM SFN and/or 100 µM Myr was assessed using an MTT assay. Apoptosis was assessed by Hoechst 33258 nuclear staining, and by detection of single‑stranded DNA using an enzyme‑linked immunosorbent assay. Compared with the effects of each compound alone, the combination of SFN and Myr synergistically reduced cell viability, induced apoptosis, increased pro‑apoptotic Bcl‑2 associated X protein expression, decreased anti‑apoptotic B‑cell lymphoma‑2 expression, enhanced Bcl‑2‑associated death promoter (Bad) translocation from the cytoplasm to the mitochondria, and reduced Bad phosphorylation at Ser112. These effects were accompanied by increased cleavage of caspase 3 and poly‑ADP‑ribose‑polymerase. In addition, combined SFN and Myr treatment significantly decreased the protein expression levels of phosphorylated AKT serine/threonine kinase 1 (Akt) at Ser473, as well as the phosphorylation of the downstream protein ribosomal protein, S6 kinase β‑1. Therefore, SFN plus Myr was a more potent inducer of apoptosis in 3T3‑L1 adipocytes than either compound alone. The results of the present study suggest that the mechanism of SNF/Myr‑induced apoptosis involved activation of the Akt‑mediated mitochondrial apoptotic pathway. This may aid treatment of animal models of obesity and preclinical testing.
Tangeretin induces the expressions of apoptotic proteins Influence of tangeretin on the expression of proteins of the apoptotic pathway was assessed by western blotting. Caspases play a crucial role as regulators of apoptosis (Zhuo et al., 2009). Tangeretin exposure bought a multi-fold increase in the expression of caspase-3. Further, as the proteins of Bcl-2 family are important in the induction of apoptosis, we also examined the effects of tangeretin on the expressions of Bcl-2 proteins. Tangeretin dose-dependently caused upregulation in the expression of Bax and Bad with a significant (p<0.05) down-regulation of anti-apoptotic proteins- Bcl-xL and Bcl-2. Tangeretin at 100 µM caused nearly two-fold enhanced expression of Bax in comparison with cells which are not exposed.
DOWNREGULATES AKT / PI3K
Combination of innocuous dietary components with anticancer drugs is an emerging new strategy for cancer chemotherapy to increase anti-tumor responses. Tangeretin (TG) is a citrus flavonoid known to inhibit cancer cell proliferation. Here, we show an enhanced response of A2780/CP70 and 2008/C13 cisplatin-resistant human ovarian cancer cells to various combination treatments of cisplatin (Cis) and tangeretin. Pretreatment of cells with tangeretin prior to cisplatin treatment synergistically inhibited cancer cell proliferation. This combination was effective in activating apoptosis via caspase cascade as well as arresting cell cycle at G2/M-phase. Moreover, phospho-Akt and its downstream substrates, e.g., NF-κB, phospho-GSK-3β and phospho-BAD were down-regulated upon tangeretin-cisplatin treatment. The tangeretin-cisplatin induced apoptosis in A2780/CP70 cells was increased by phosphoinositide-3 kinase (PI3K) inhibition and siRNA-mediated Akt silencing, but reduced by over-expression of constitutively activated-Akt and GSK-3β inhibition. The overall results indicated that tangeretin exposure preconditions cisplatin-resistant human ovarian cancer cells for a conventional response to low-dose cisplatin-induced cell death occurring through down-regulation of PI3K/Akt signaling pathway. Thus, effectiveness of tangeretin combinations, as a promising modality in the treatment of resistant cancers, warrants systematic clinical studies.
CDK2 CDK4 INHIBITOR / P21 P27 Activation
Tangeretin (5,6,7,8,4’-pentamethoxyflavone) is concentrated in the peel of citrus fruits. DNA flow cytometric analysis indicated that tangeretin blocked cell cycle progression at G1 phase in colorectal carcinoma COLO 205 cells. Over a 24 h exposure to tangeretin, the degree of phosphorylation of Rb was decreased after 12 h and G1 arrest developed. The protein expression of cyclins A, D1, and E reduced slightly under the same conditions. Immunocomplex kinase experiments showed that tangeretin inhibited the activities of cyclin-dependent kinases 2 (Cdk2) and 4 (Cdk4)in a dose-dependent manner in the cell-free system. As the cells were exposed to tangeretin (50 μM) over 48 h a gradual loss of both Cdk2 and 4 kinase activities occurred. Tangeretin also increased the content of the Cdk inhibitor p21 protein and this effect correlated with the elevation in p53 levels. In addition, tangeretin also increased the level of the Cdk inhibitor p27 protein within 18 h. These results suggest that tangeretin either exerts its growth-inhibitory effects through modulation of the activities of several key G1 regulatory proteins, such as Cdk2 and Cdk4, or mediates the increase of Cdk inhibitors p21 and p27.
In summary, we have shown that the growth-inhibitory response to tangeretin, including the inhibition of Cdk2 and Cdk4 kinase activities, and the increase in the level of p21 and p27,is dependent on p53 activity. The p21 and/or p27 up-regulation by tangeretin might play an important role in its anti-proliferative activity in cultured cells. A predominance of polymethoxyflavonoids (tangeretin and nobiletin) over polyhydroxyflavonoids was demonstrated by the induced p53 levels (Figure 8). In previous reports polymethoxyflavonoids were more potent than polyhydroxyflavonoids in antitumor activities using a cell culture system (42). Differences in their activities may be derived from the relatively greater membrane uptake efficiencies of polymethoxyflavonoids than those of polyhydoxyflavonoids because methylation of the phenolic hydroxyl groups, in general, increases the molecular hydrophobicity that promotes the transportation rates (5). To our knowledge this is the first report to demonstrate that tangeretin does, indeed, play an important role in the function of anti-proliferation and antitumor capacity through inhibiting Cdks kinase activities and elevating Cdk inhibitors, and may provide a pivotal mechanism for its cancer chemopreventive action.
The main source of tangeretin is citrus peel 69 and thus tangeretin is categorized as a citrus flavonoid. A paper published in 2007 reported that tangeretin suppressed proliferation by cell cycle arrest in G1 without apoptosis in the human colon cancer cell line HT-29 70. Multidrug-resistant (LoVo/Dx) human colon adenocarcinoma cells that were exposed to tangeretin were inhibited; the investigators concluded that the activity of tangeretin probably enhanced apoptosis activity through the induction of caspase-3 in this cell line 71. Tangeretin suppresses the proliferation of COLO-205 by blocking cell cycle progression in G1; reducing the expression of cyclin A, D, and E; reducing the effect of CDK2 and CDK4; and stimulating the activity of p21, p27, and p53 72.

TaraxasterolTrichostatin A
upregulates Hint1, Bax / downregulates Bcl2, cyclin D1
Taraxasterol has potent anti-inflammatory and anti-tumor activity. However, the effect and potential mechanisms of Taraxasterol on the growth of human liver cancer have not been clarified. Histidine triad nucleotide-binding protein 1 (Hint1) is a tumor suppressor and its downregulated expression is associated with the development of cancer. Here, we report that Taraxasterol treatment significantly suppressed cell proliferation and induced cell cycle arrest at G0/G1 phase and apoptosis in liver cancer cells, but not in non-tumor hepatocytes. Furthermore, Taraxasterol upregulated Hint1 and Bax, but downregulated Bcl2 and cyclin D1 expression, accompanied by promoting the demethylation in the Hint1 promoter region in liver cancer cells. The effects of Taraxasterol were abrogated by Hint1 silencing and partially mitigated by Bax silencing, Bcl2 or cyclin D1 over-expression in HepG2 cells. Moreover, oral administration with Taraxasterol did not affect body weight, urinary protein levels, and the heart, liver, and kidney morphology in BALB/c mice but effectively inhibited the growth of implanted SK-Hep1 tumor in vivo. Collectively, we demonstrate that Taraxasterol inhibits the growth of liver cancer at least partially by enhancing Hint1 expression to regulate Bax, Bcl2, and cyclin D1 expression. Taraxasterol may be a drug candidate for the treatment of human liver cancer.
POTENT BCL, 2, BCL-XL, BCL-W, MCL-1 INHIBITOR
Taraxasterol shows highest mean binding affinity for Bcl-2 family proteins. Taraxasterol also shows highest individual binding affinity for Bcl-XL protein. Taraxasterol, a pentacyclic-triterpene was isolated from various medicinal herbs, is well
known for anti-inflammatory, anti-asthmatic activity and also various effects like cholesterol-lowering, anti-microbial, anti- bacterial, anti-fungal effects etc. Ability to induce apoptosis in drug resistant cancers, chemoprotective/ chemopreventive is also reported in literature (Ovesna et al., 2004).
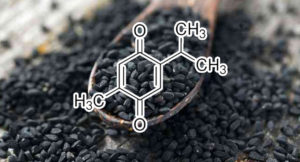
Thymoquinone
(from Nigella sativa aka “black caraway” and “black cumin”)
down‐regulateS STAT3, Bcl‐2, Bcl‐xL, survivin
Constitutive activation of the signal transducer and activator of transcription 3 (STAT3) pathway is frequently encountered in several human cancers including multiple myeloma (MM). Thus, agents that suppress STAT3 phosphorylation have a potential for treatment of MM. In the present report, we investigated whether thymoquinone (TQ), the main component isolated from the medicinal plant Nigella sativa, modulated the STAT3 signalling pathway in MM cells.
The effect of TQ on both constitutive and IL‐6‐induced STAT3 activation, associated protein kinases, STAT3‐regulated gene products involved in proliferation, survival and angiogenesis, cellular proliferation and apoptosis in MM cells, was investigated.
KEY RESULTS We found that TQ inhibited both constitutive and IL‐6‐inducible STAT3 phosphorylation which correlated with the inhibition of c‐Src and JAK2 activation. Vanadate reversed the TQ‐induced down‐regulation of STAT3 activation, suggesting the involvement of a protein tyrosine phosphatase. Indeed, we found that TQ can induce the expression of Src homology‐2 phosphatase 2 that correlated with suppression of STAT3 activation. TQ also down‐regulated the expression of STAT3‐regulated gene products, such as cyclin D1, Bcl‐2, Bcl‐xL, survivin, Mcl‐1 and vascular endothelial growth factor. Finally, TQ induced the accumulation of cells in sub‐G1 phase, inhibited proliferation and induced apoptosis, as indicated by poly ADP ribose polymerase cleavage. TQ also significantly potentiated the apoptotic effects of thalidomide and bortezomib in MM cells.
Our study has identified STAT3 signalling as a target of TQand has thus raised its potential application in the prevention and treatment of MM and other cancers.
Purpose: In this study, we have investigated the effect of trichostatin A (TSA) pretreatment on the cytotoxicity of TRAIL in TRAIL-resistant myeloma cells. Methods and Results: MM1S myeloma cells exhibited resistance to TRAIL-induced apoptosis even at high doses of TRAIL and was sensitive to low doses of TSA. Sequential treatment of myeloma cells with TSA followed by TRAIL enhanced TRAIL cytotoxicity. TSA induced the transcription of TRAIL death receptor DR5 without affecting the transcription of DR4 and the decoy receptors; DcR1 and DcR2. However, the surface expression of both DR4 and DR5 was not modulated by TSA treatment. TSA treatment repressed the transcription and downregulated the expression of the antiapoptotic Bcl-2 proteins; Bcl-2 and Bcl-XL. Surprisingly, the effect of TSA on the proapoptotic Bcl-2 proteins was mixed, the two isoforms of PUMA (α, β), Noxa, Bax were downregulated, while Bim was upregulated. Although MM1S cells showed higher expression level of FLIPS than other TRAIL-sensitive myeloma cells, the enhancing effect of TSA was not accompanied by FLIPSdownregulation. Conclusion: In conclusion, TSA sensitized TRAIL-resistant myeloma cells by downregulating the antiapoptotic Bcl-2 protein without altering FLIPS expression.
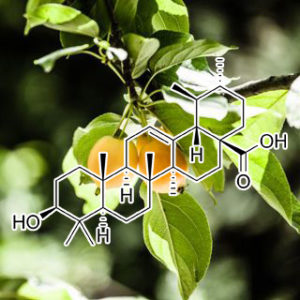
Ursolic AcID
USP7 INHIBITOR
Deubiquitinating protease USP7 is a promising therapeutic target for cancer treatment, and interest in developing USP7 inhibitors has greatly increased. In the present study, we reported a series of natural pentacyclic triterpenes with USP7 inhibitory activity in vitro. Among them, both the ursane triterpenes and oleanane triterpenes were more active than the lupine triterpenes, whereas ursolic acid was the most potent with IC50 of 7.0±1.5 μmol/L. Molecular docking studies showed that ursolic acid might occupy the ubiquitin binding pocket of USP7, with the 17-carboxyl group and 3-hydroxyl group playing a vital role in the USP7-ursolic acid interaction. Using the cellular thermal shift assay, we demonstrated that ursolic acid interacted with USP7 in RPMI8226 human myeloma cells. Ursolic acid dose-dependently inhibited the proliferation of the myeloma cells with IC50 of 6.56 μmol/L, accompanied by reductions in USP7 substrates such as MDM2, UHRF1 and DNMT1. Overexpression of USP7 partially, but significantly attenuated ursolic acid-induced cell death as well as downregulation of MDM2, UHRF1 and DNMT1. In conclusion, we demonstrate for the first time that pentacyclic triterpenes represent a novel scaffold for developing USP7 inhibitors and that USP7 inhibition contributes to the anti-cancer effect of ursolic acid.
STAT3 INHIBITOR
The signal transducer and activator of transcription 3 (STAT3) has been found to be constitutively active in liver cancer. There is no STAT3 inhibitors approved to be used clinically for the treatment or prevention of liver cancer. Some dietary compounds including ursolic acid (UA) have been reported to inhibit the growth of cancer cells. However, whether UA could inhibit STAT3 phosphorylation in hepatocellular carcinoma has not been reported. The inhibitory effects of UA on STAT3 phosphorylation, along with cell viability, migration, colony formation in vitro, as well as tumor growth in vivo were examined in human liver cancer cell lines. Our data showed that UA inhibited the P-STAT3 induced by interleukin-6 (IL-6) in Hep3B liver cancer cells which express very low basal level of P-STAT3. The constitutive STAT3 phosphorylation was also inhibited by UA in HEPG2, 7721 and Huh7 human liver cancer cell lines. UA decreased the expression of downstream target genes of STAT3, such as Bcl-2, Bcl-xl and survivin in general, with difference in these cell lines. UA also suppressed cell viability, cell migration and colony formation in liver cancer cells. Furthermore, UA suppressed STAT3 phosphorylation and HEPG2 tumor growth by oral daily treatment in vivo. UA, which exists widely in fruits and herbs, could inhibit STAT3 activation and the growth of human liver cancer cells in vitro and in vivo. It might be a potential health care product that could be used daily for prevention, as well as a promising candidate for chemotherapy of liver cancer.
Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice
Activation of transcription factors nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) is frequently observed in prostate cancer and has been linked with tumor cell proliferation, invasion, metastasis, and angiogenesis. In this study, we investigated the effect of ursolic acid (UA) on NF-κB and STAT3 signaling pathways in both androgen-independent (DU145) and androgen-dependent (LNCaP) prostate cancer cell lines and also prospectively tested the hypothesis of NF-κB and STAT3 inhibition using a virtual predictive functional proteomics tumor pathway technology platform. We found that UA inhibited constitutive and TNF-α-induced activation of NF-κB in DU145 and LNCaP cells in a dose-dependent manner. The suppression was mediated through the inhibition of constitutive and TNF-α-induced IκB kinase (IKK) activation, phosphorylation of IκBα and p65 and NF-κB-dependent reporter activity. Furthermore, UA suppressed both constitutive and inducible STAT3 activation in prostate cancer cells concomitant with suppression of activation of upstream kinases (Src and JAK2) and STAT3-dependent reporter gene activity. UA also downregulated the expression of various NF-κB and STAT3 regulated gene products involved in proliferation, survival, and angiogenesis and induced apoptosis in both cells lines as evidenced by DNA fragmentation and annexin V staining. In vivo, UA (200 mg/kg b.w.) treated for 6 weeks inhibited the growth of DU145 cells in nude mice without any significant effect on body weight. Overall, our results from experimental and predictive studies suggest that UA mediates its anti-tumor effects through suppression of NF-κB and STAT3 pathways in prostate cancer.
CYCLIN D1 INHIBITOR
UA treatment decreased cyclin D1, pERK1/2, and Cullin1 levels in a dose- and time-dependent manner and UA markedly inhibited FBXW8. Treatment of HEC108 cells moderately decreased Rbx1 in a dose- and-time-dependent fashion. In contrast, UA treatment increased ubiquitinated proteins in a dose- and time-dependent manner in both cell lines. RING-type E3 ligase accumulated in the cytoplasm following UA treatment of SNG-2cells. That in turn prevented cytoplasmic degradation of cyclin D1 via RING-type E3 (SCF E3s) ligase. In conclusion, our study found inhibition of the MAPK- cyclin D1 pathway and RING type E3 ligase (SCF E3s) in both endometrial cancer cell lines. Furthermore, CD36 was noted as a cell surface receptor for UA.
Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21waf1 and NOXA in pancreatic cancer cells
A series of new heterocyclic derivatives of ursolic acid 1 were synthesized and evaluated for their antiproliferative activity against AsPC-1 pancreatic cancer cells. Compounds 24–32, with an α,β unsaturated ketone in conjugation with an heterocyclic ring in ring A have improved antiproliferative activities. Compound 32 is the most active compound with an IC50 of 1.9 μM which is sevenfold more active than ursolic acid 1. Compound 32 arrests cell cycle in G1 phase and induces apoptosis in AsPC-1 cells with upregulation of p53, p21waf1 and NOXA protein levels.
Urolithin A
Urolithins are gut microbiota metabolites of ellagitannins; ellagi- tannin-rich foods include walnuts, red raspberries, strawberries, and pomegranates. Since urolithin A (UA) and B are able to attenuate LPS- induced inflammation, inhibiting activation of the NF-κB and Akt sig- naling pathways (Xu et al., 2018), they may emerge as new anti-SASP metabolites. It has recently been noted that oral UA can induce mitophagy both in vitro and in vivo. In C. elegans, UA prevented age-related accumulation of dysfunctional mitochondria and extended lifespan, whereas in rodents it improved exercise capacity both in different mouse models of age-related decline of muscle function and in young rats (Ryu et al., 2016).The anti-SASP and senolytic effects of synthetic and natural com- pounds.
EXTENDS LIFESPAN
Urolithin A is one of the three end-products (the other two being Urothilin B and C) of microflora-mediated processing of ellagic acid in the gut from ellagitannins. It is contained in berries, acorns, nuts and tree leaves. When fed to C. elegans, urolithin A extends lifespan and improves healthspan (Ryu et al., 2016), and reduces Aβ-associated memory loss (Fang et al., 2019). The lifespan-extending properties of urolithin A are at least partially dependent on AMPK, and completely dependent on intact mitochondria, as it induces mitophagy (Ryu et al., 2016). The worm lifespan extension produced by urolithins was completely dependent on Pink1, dct-1/BNIP3, Beclin, pdr-1/Parkin, sqst-1, vps-34 and skn-1/Nrf1/2. In mice, urolithin A treatment has also been linked to mitophagy induction and improved muscle function (Ryu et al., 2016).
NF-κB↓, PARP↑, IIα↓, p-p53↑, caspase-3↑, caspase-7↑, c-JNK↑, PKCε↓, IKKα↓, Bax↑, Bcl- xL↓, p21↑, p27↑, Bcl-2↓, IL6↓, IL6R↓, c-myc, IKK↓, p-TAK1, IKKβ↓, IKKα↓, IL1β↓, STAT-3↓, TLR-4↑, TLR-7↑, TLR-8↑, Akt↓, TNFα↓, IκB↓
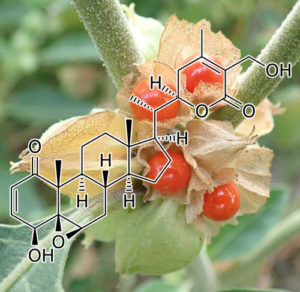
Withaferin A (Ashwaganda)
(from Withania somnifera aka “Indian Winter cherry” or “Ashwagandha” in Sanskrit)
Key Functions:
HSP90 InhibitorHSPs (Heat shock proteins) are highly conserved ubiquitous proteins among species which are involved in maintaining appropriate folding and conformation of other proteins and are thus referred to as molecular chaperones. Hsp90 (Heat-shock protein 90 kDa) is one of a group of molecular chaperones responsible for managing protein folding and quality control in cell environment. However it is also involved in the maturation and stabilization of a wide range of oncogenic client proteins which are crucial for oncogenesis and malignant progression. Hsp90 requires a series of co-chaperones to assemble into a super-chaperone complex for its function. These co-chaperones bind and leave the complex at various stages to regulate the chaperoning process. Arresting the chaperone cycle at these stages by targeting different co-chaperone/Hsp90 interactions seems to be quite a viable alternative and is likely to achieve similar consequences as that of Hsp90 direct inhibition with added favors of high specificity and reduced side effect profile. The study conducted here is an attempt to explore the potential of Withania somnifera’s major constituent WA (Withaferin A) in attenuating the Hsp90/Cdc37 chaperone/co-chaperone interactions for enhanced tumor arresting activity and to elucidate the underlying mode of action using computational approaches.
BCL INHIBITORAn active medicinal component of plant origin with an ability to overcome autophagy by inducing apoptosis should be considered a therapeutically active lead pharmacophore to control malignancies. In this report, we studied the effect of concentration-dependent 3-AWA (3-azido withaferin A) sensitization to androgen-independent prostate cancer (CaP) cells which resulted in a distinct switching of 2 interrelated conserved biological processes, i.e. autophagy and apoptosis. We have observed 3 distinct parameters which are hallmarks of autophagy in our studies. First, a subtoxic concentration of 3-AWA resulted in an autophagic phenotype with an elevation of autophagy markers in prostate cancer cells. This led to a massive accumulation of MAP1LC3B and EGFP-LC3B puncta coupled with gradual degradation of SQSTM1. Second, higher toxic concentrations of 3-AWA stimulated ER stress in CaP cells to turn on apoptosis within 12 h by elevating the expression of the proapoptotic protein PAWR, which in turn suppressed the autophagy-related proteins BCL2 and BECN1. This inhibition of BECN1 in CaP cells, leading to the disruption of the BCL2-BECN1 interaction by overexpressed PAWR has not been reported so far. Third, we provide evidence that pawr-KO MEFs exhibited abundant autophagy signs even at toxic concentrations of 3-AWA underscoring the relevance of PAWR in switching of autophagy to apoptosis. Last but not least, overexpression of EGFP-LC3B and DS-Red-BECN1 revealed a delayed apoptosis turnover at a higher concentration of 3-AWA in CaP cells. In summary, this study provides evidence that 3-AWA is a strong anticancer candidate to abrogate protective autophagy. It also enhanced chemosensitivity by sensitizing prostate cancer cells to apoptosis through induction of PAWR endorsing its therapeutic potential.
Wogonin
Wogonin suppresses tumor growth in vivo and VEGF-induced angiogenesis through inhibiting tyrosine phosphorylation of VEGFR2.
Previous studies revealed that wogonin, a naturally occurring monoflavonoid extracted from Scutellariae radix, possessed anticancer activity both in vitro and in vivo. However, the molecular mechanism of its potent anticancer activity remains poorly understood and warrants further investigations. In this study, we found for the first time that wogonin inhibited the growth and tumor angiogenesis of human gastric carcinoma in nude mice. We explored the inhibitory effect of wogonin on angiogenesis stimulated by vascular endothelial growth factor (VEGF) in vitro. Wogonin suppressed the VEGF-stimulated migration and tube formation of human umbilical vein endothelial cells (HUVECs). It also restrained VEGF-induced tyrosine phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2). This inhibition of receptor phosphorylation was correlated with a significant decrease in VEGF-triggered phosphorylated forms of ERK, AKT and p38. Taken together, these findings strongly suggest that wogonin might be a promising antitumor drug.
MDM2 INHIBITOR
Most cancer cells generate energy through aerobic glycolysis to enable their rapid growth and proliferation, which is a phenomenon known as Warburg effect. Inhibition of aerobic glycolysis reduces lactate and ATP generation in cancer cells, and ultimately kills tumor cells. Increasing evidence suggests that wogonin, a flavonoid isolated from Scutellaria baicalensis Georgi, exhibits potent anti-tumor effects in vivo and in vitro. However, the role of wogonin in the aerobic glycolysis of tumor cells has not yet been elucidated. In this study, the effect of wogonin on glucose uptake, lactate generation and ATP content is assessed in colon, ovarian and hepatocellular cancer cells. The results indicate that wogonin reduces glycolysis and cell proliferation in cancer cells expressing wild-type p53 but not mutated p53. Wogonin increases the expression of p53 and p53-inducible glycolysis and apoptosis regulator (TIGAR), while decreases glucose transporter 1 (GLUT1) and some key glycolytic enzymes. Expressing wild-type and mutant-type p53 in HCT116 p53−/− cells proved that the inhibitory effect of wogonin on glycolysis in cancer cells is dependent on wild type p53. Mechanistically, wogonin induced the phosphorylation and acetylation of p53 and inhibited the expression of MDM2 to enhance the stability of p53. Furthermore, wogonin suppressed the growth and glycolysis of transplanted wild-type p53 expressing A2780 cells on nude mice, but did not affect mutant-type p53 expressing HT-29 cells. In conclusion, these findings explain the broad anti-tumor effect of wogonin, and offer a novel avenue for the therapeutic strategy in cancer.
Wogonin and Related Natural Flavones Overcome Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL) Protein Resistance of Tumors by Down-regulation of c-FLIP
Background: Most primary cancer cells are resistant to TRAIL-mediated apoptosis.
Results: Wogonin and related natural flavones suppress c-FLIP but up-regulate TRAIL receptor-2 expression and thereby sensitize resistant tumor cells to TRAIL-mediated apoptosis.
Conclusion: Wogonin and related natural flavones can overcome TRAIL resistance of cancer cells.
Significance: These flavones can be developed as adjuvants for TRAIL-based anticancer therapy.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising anticancer agent that kills various tumor cells without damaging normal tissues. However, many cancers remain resistant to TRAIL. To overcome TRAIL resistance, com- bination therapies using sensitizers of the TRAIL pathway would be an efficacious approach. To investigate potential sensitizers of TRAIL-induced apoptosis, we used TRAIL-resistant human T cell leukemia virus type 1 (HTLV-1)-associated adult T cell leukemia/ lymphoma (ATL) cells as a model system. So far, HTLV-1-associ- ated ATL is incurable by presently known therapies. Here, we show that wogonin and the structurally related natural flavones apigenin and chrysin break TRAIL resistance in HTLV-1-associated ATL by transcriptional down-regulation of c-FLIP, a key inhibitor of death receptor signaling, and by up-regulation of TRAIL receptor 2 (TRAIL-R2). This effect is mediated through transcriptional inhi- bition of the p53 antagonist murine double minute 2 (Mdm2), leading to an increase in p53 levels and, consequently, to up-regulation of the p53 target gene TRAIL-R2. We also show that these flavones can sensitize to TNF- and CD95-mediated cell death. Further- more, we show that wogonin, apigenin, and chrysin also enhance TRAIL-mediated apoptosis in other human cancer cell lines includ- ing breast cancer cell line MDA-MB-231, colon cancer cell line HT-29, hepatocellular carcinoma cell line HepG2, melanoma cell line SK-MEL-37, and pancreatic carcinoma cell line Capan-1 by the same mechanism. Thus, our study suggests the potential use of these flavones as an adjuvant for TRAIL-mediated anticancer therapy.
CASPASE 3 ACTIVATOR; MCL-1 Inhibitor
Seven structurally related flavonoids including luteolin, nobiletin, wogonin, baicalein, apigenin, myricetin and fisetin were used to study their biological activities on the human leukemia cell line, HL-60. On MTT assay, wogonin, baicalein, apigenin, myricetin and fisetin showed obvious cytotoxic effects on HL-60 cells, with wogonin and fisetin being the most-potent apoptotic inducersamong them. The cytotoxic effects of wogonin and fisetin were accompanied by the dose- and time-dependent appearance of characteristics of apoptosis including DNA fragmentation, apoptotic bodies and the sub-G1 ratio. Treatment with an apoptosis-inducing concentration of wogonin or fisetin causes rapid and transient induction of caspase 3/CPP32 activity, but not caspase 1 activity. Further, cleavage of poly(ADP-ribose) polymerase (PARP) and decrease of pro-caspase 3 protein were detected in wogonin- and fisetin-treated HL-60 cells. An increase in the pro-apoptotic protein, bax, and a decrease in the anti-apoptotic protein, Mcl-1, were detected in fisetin- and wogonin-treated HL-60 cells. However, Bcl-2, Bcl-XL, and Bad all remained unchanged in wogonin- and fisetin-treated HL-60 cells. In vitro chromatin digestion revealed that endonuclease activity was profoundly enhanced in wogonin- and fisetin-treated HL-60 cells, and the addition of ethylenediaminetetraacetic acid (EDTA) or ethyleneglycoltetraacetic acid (EGTA) into the reaction blocked endonuclease activation and at an optimum pH of 7.5. The caspase 3 inhibitor, Ac-DEVD-CHO, but not the caspase 1 inhibitor, Ac-YVAD-CHO, attenuated wogonin- and fisetin-induced DNA ladders, PARP cleavage, and endonuclease activation. Pretreatment of HL-60 cells with N-acetyl-cysteine or catalase efficiently inhibited H2O2 (200 μM)-induced apoptosis, but showed no inhibitory effect on wogonin- and fisetin-induced DNA ladders, caspase 3 activation, or bax protein induction. Decrease in endogenous ROS production was detected in wogonin- and fisetin-treated HL-60 cells by DCHF-DA assay. In conclusion, our experiments indicate that a decrease in intracellular peroxide level was involved in wogonin- and fisetin-induced apoptosis; activation of caspase 3 and endonuclease, induction of bax protein and suppression of Mcl-1protein were detected in the process.
Anti-Obesity Effects of Xanthohumol Plus Guggulsterone in 3T3-L1 Adipocytes
Xanthohumol (XN) and guggulsterone (GS) have each been shown to inhibit adipogenesis and induce apoptosis in adipocytes. In the present study effects of the combination of XN + GS on 3T3-L1 adipocyte apoptosis and adipogenesis were investigated. Mature adipocytes were treated with XN and GS individually and in combination. XN and GS individually decreased cell viability, but XN + GS caused an enhanced decrease in viability and potentiated induction of apoptosis. Likewise, XN + GS caused a potentiated increase in caspase-3/7 activation, whereas neither of the compounds showed any effect individually. In addition, western blot analysis revealed that XN + GS increased Bax expression and decreased Bcl-2 expression, whereas individual compounds did not show any significant effect. XN and GS both decreased lipid accumulation. Individually, XN at 1.5 μM and GS at 3.12 μM decreased lipid accumulation by 26 ± 4.5% (P < .001) each, whereas XN1.5 + GS3.12 decreased lipid accumulation by 78.2 ± 1.8% (P < .001). Moreover, expression of the adipocyte-specific proteins was down-regulated with XN1.5 + GS3.12, but no effect was observed with the individual compounds. Finally, XN + GS caused an enhanced stimulation of lipolysis. Thus, combination of XN and GS is more potent in exerting anti-obesity effects than additive effects of the individual compounds.
Zerumbone
(a sesquiterpene from Zingiber Zerumbet Smith aka “Lempoyang” and “Southeast Asian Ginger”)





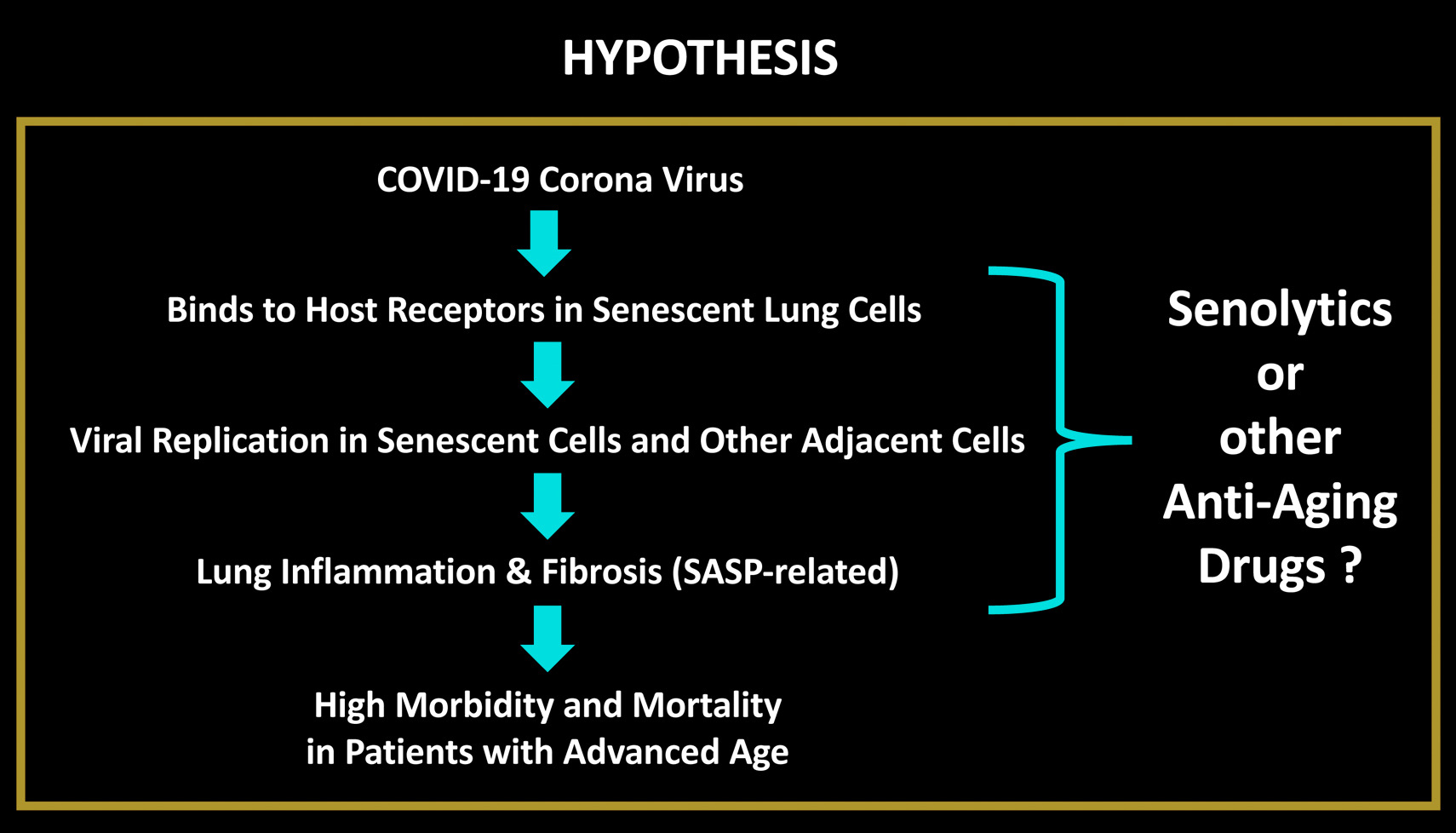








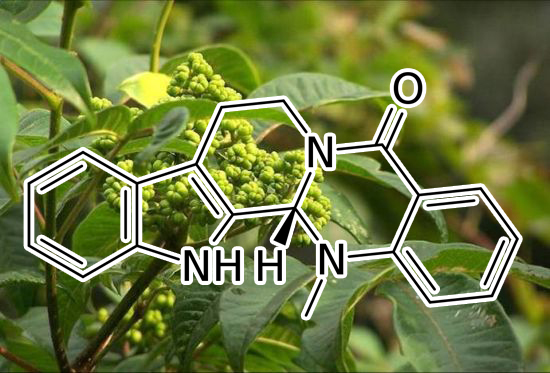
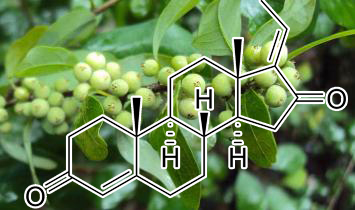

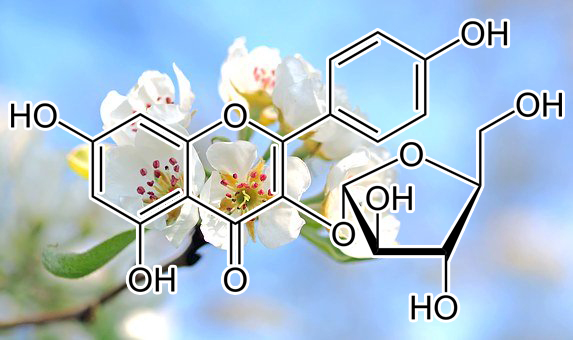

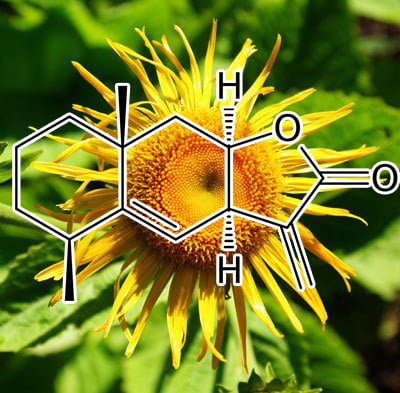
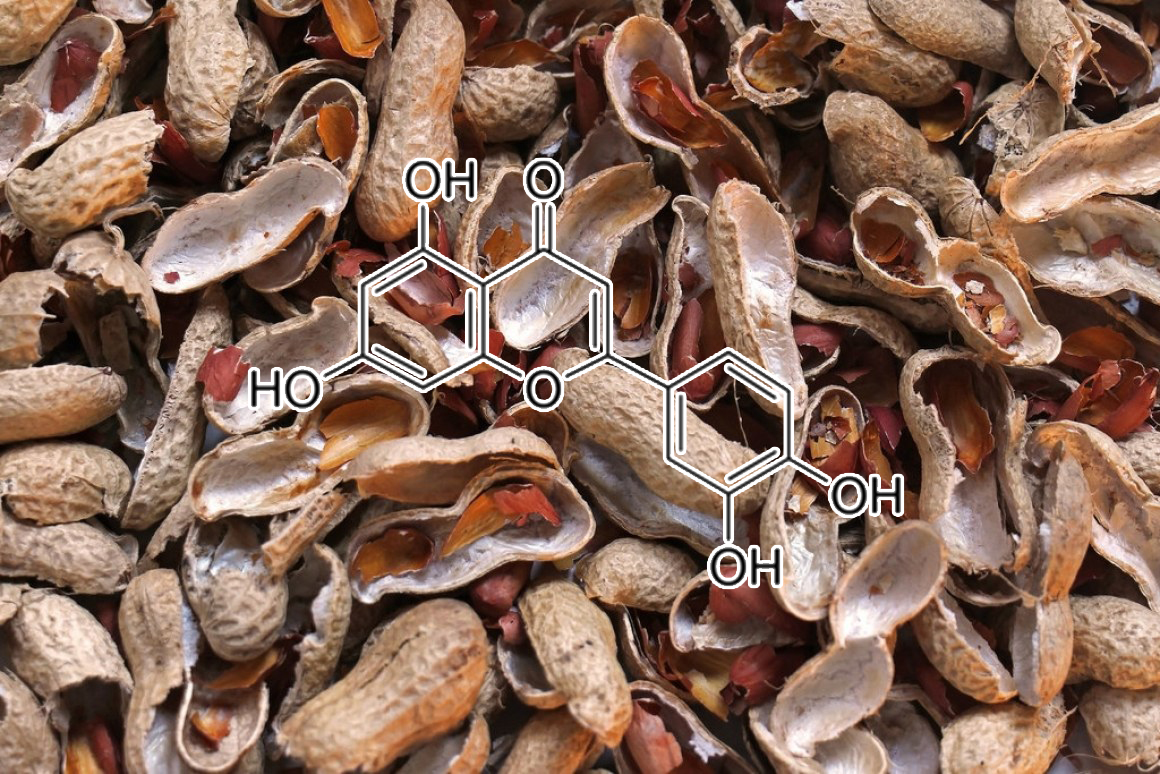



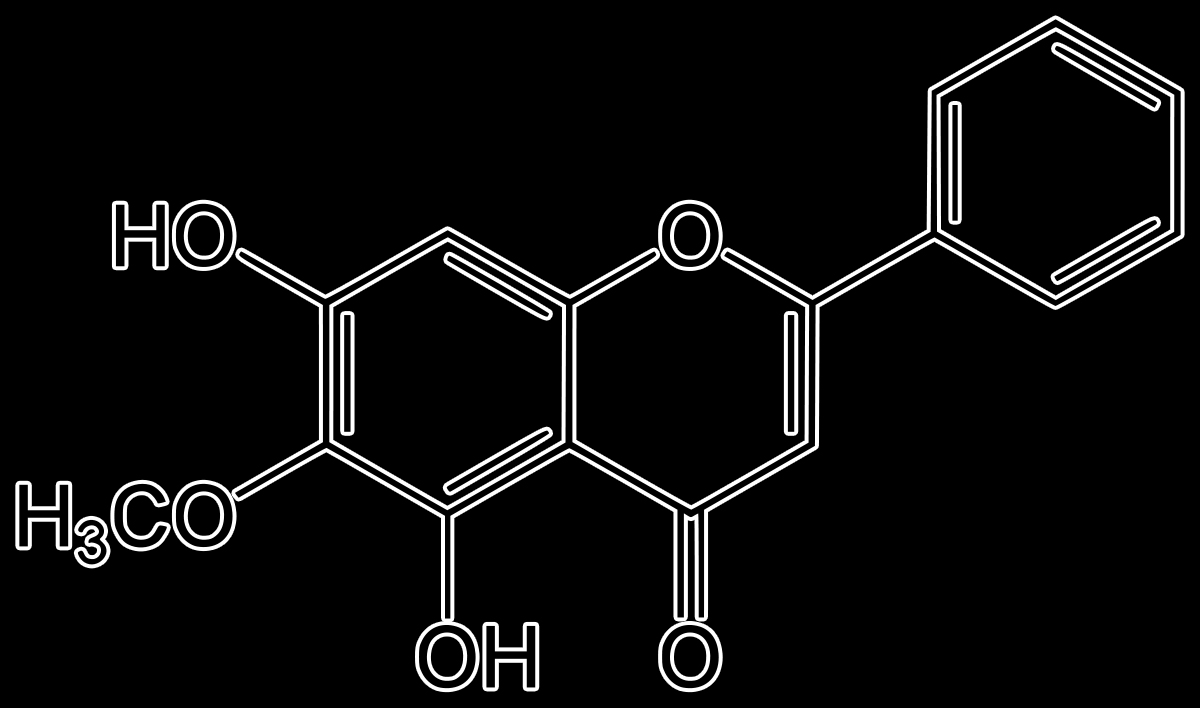
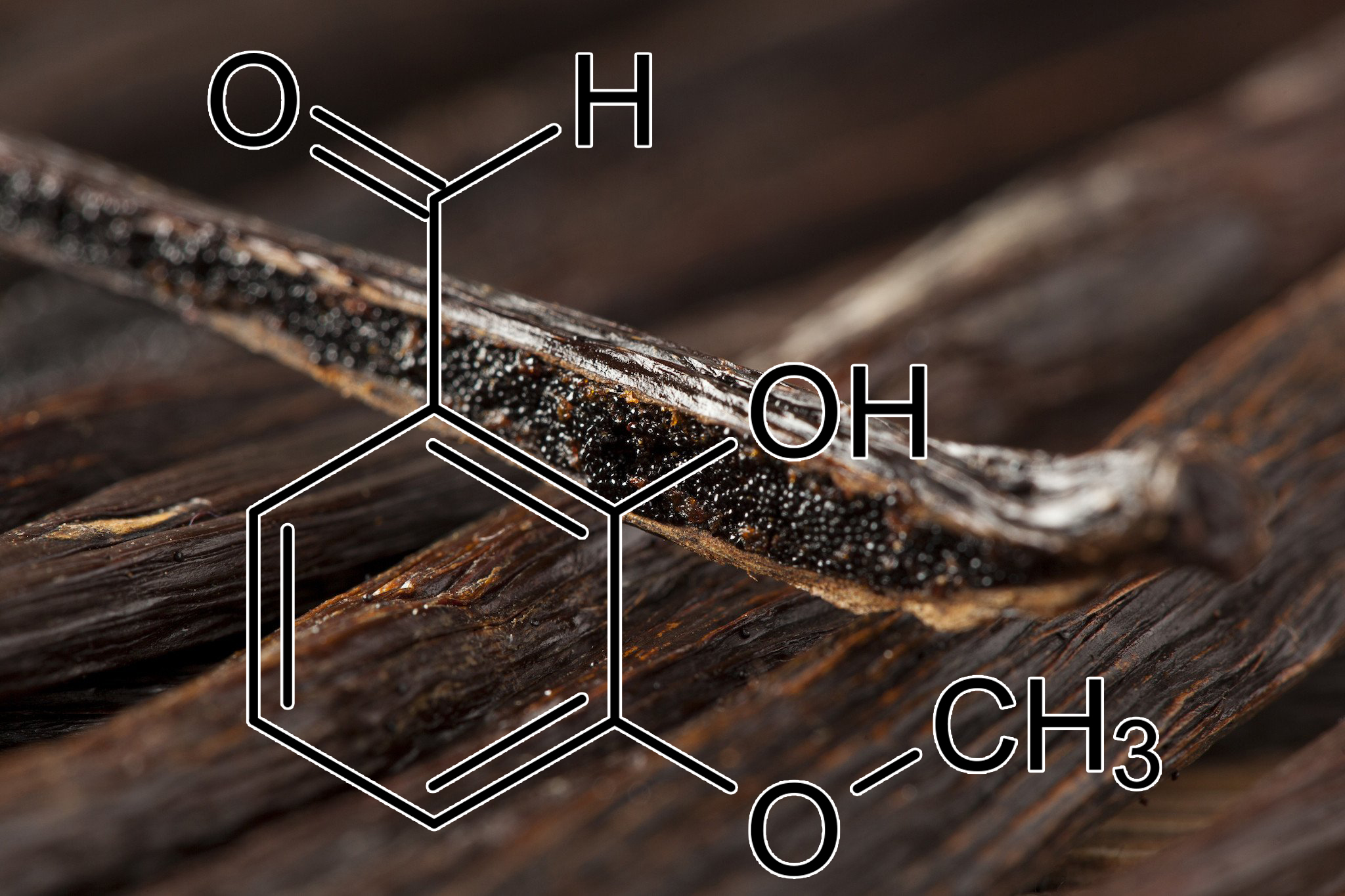
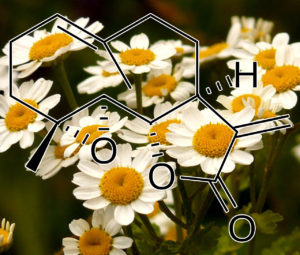

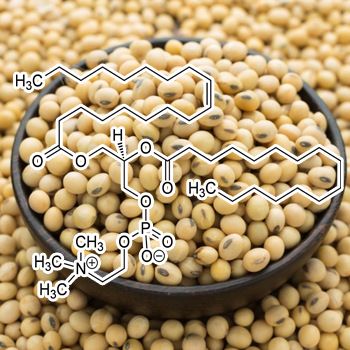


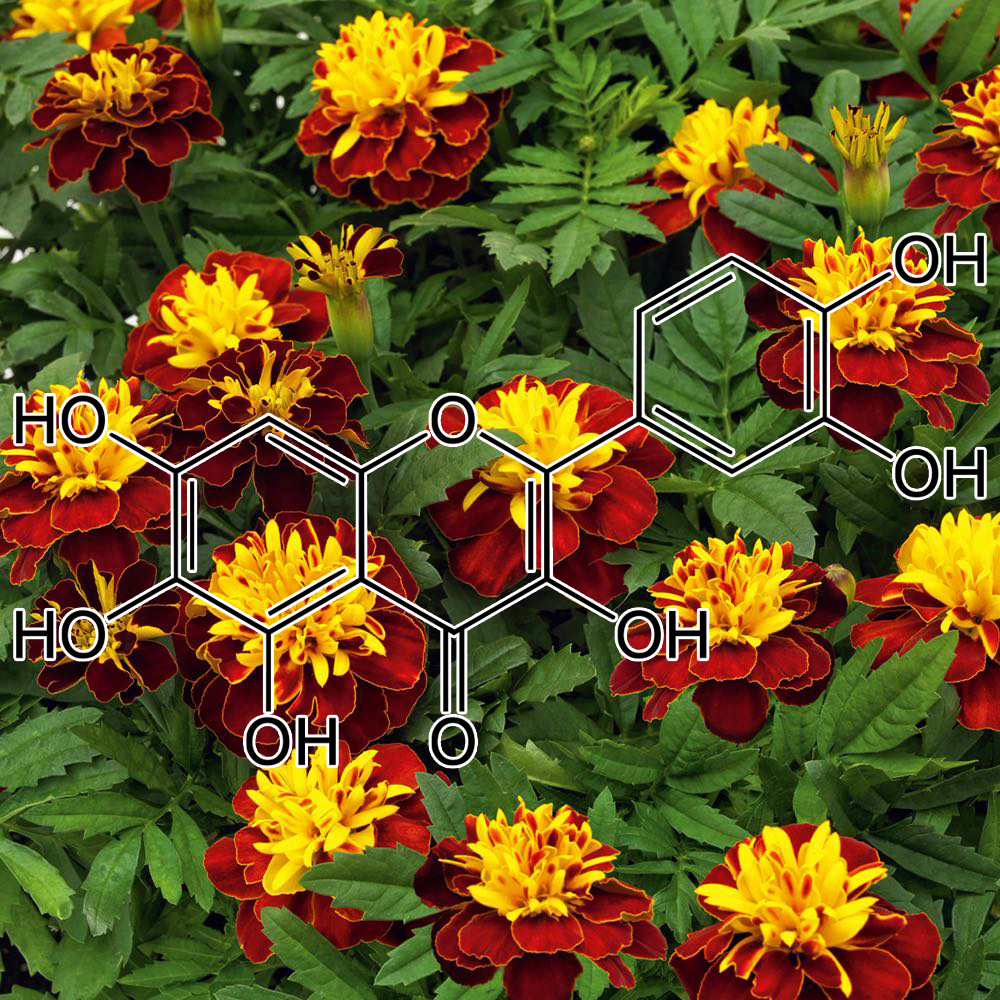

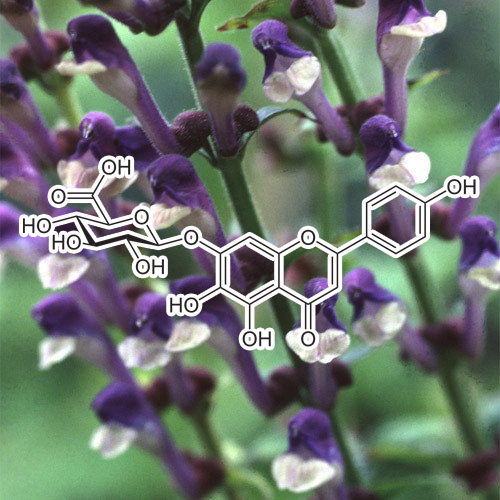
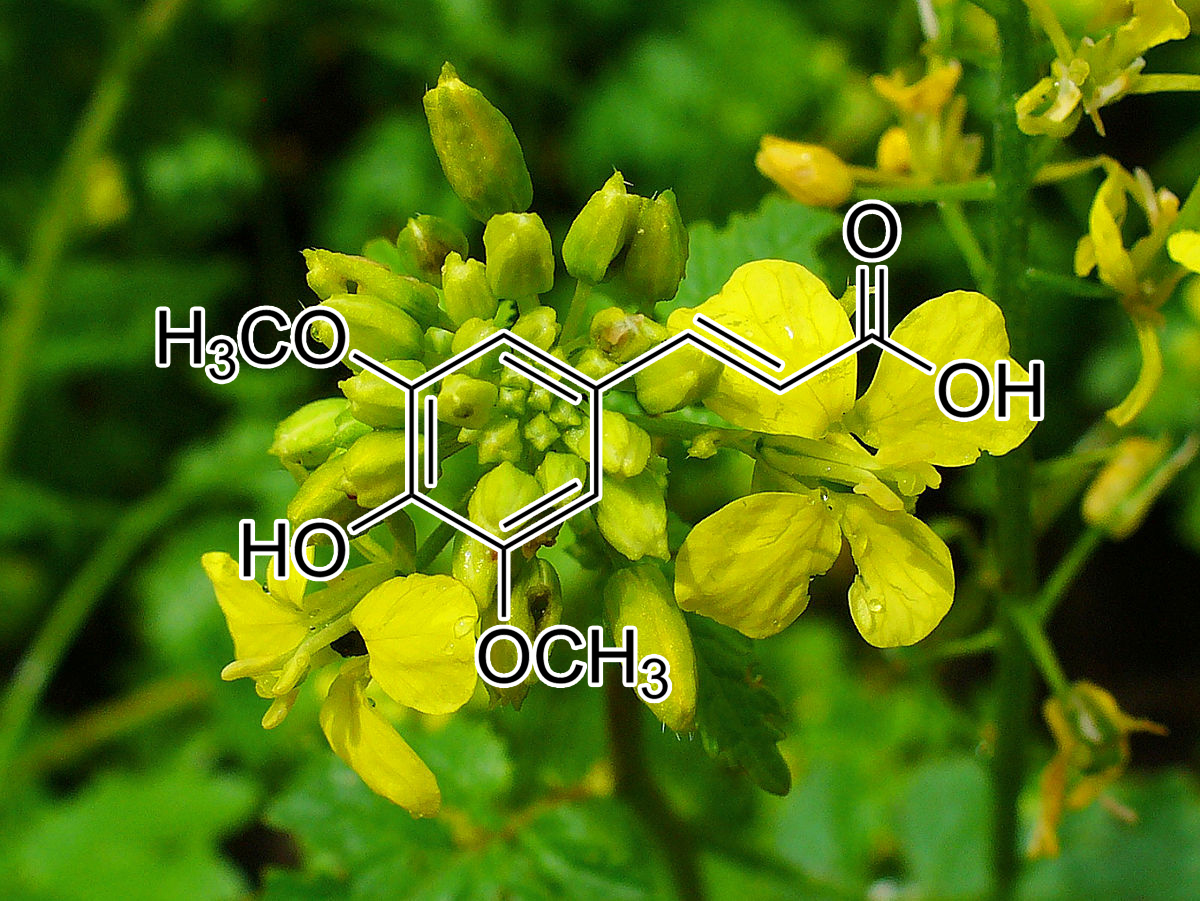






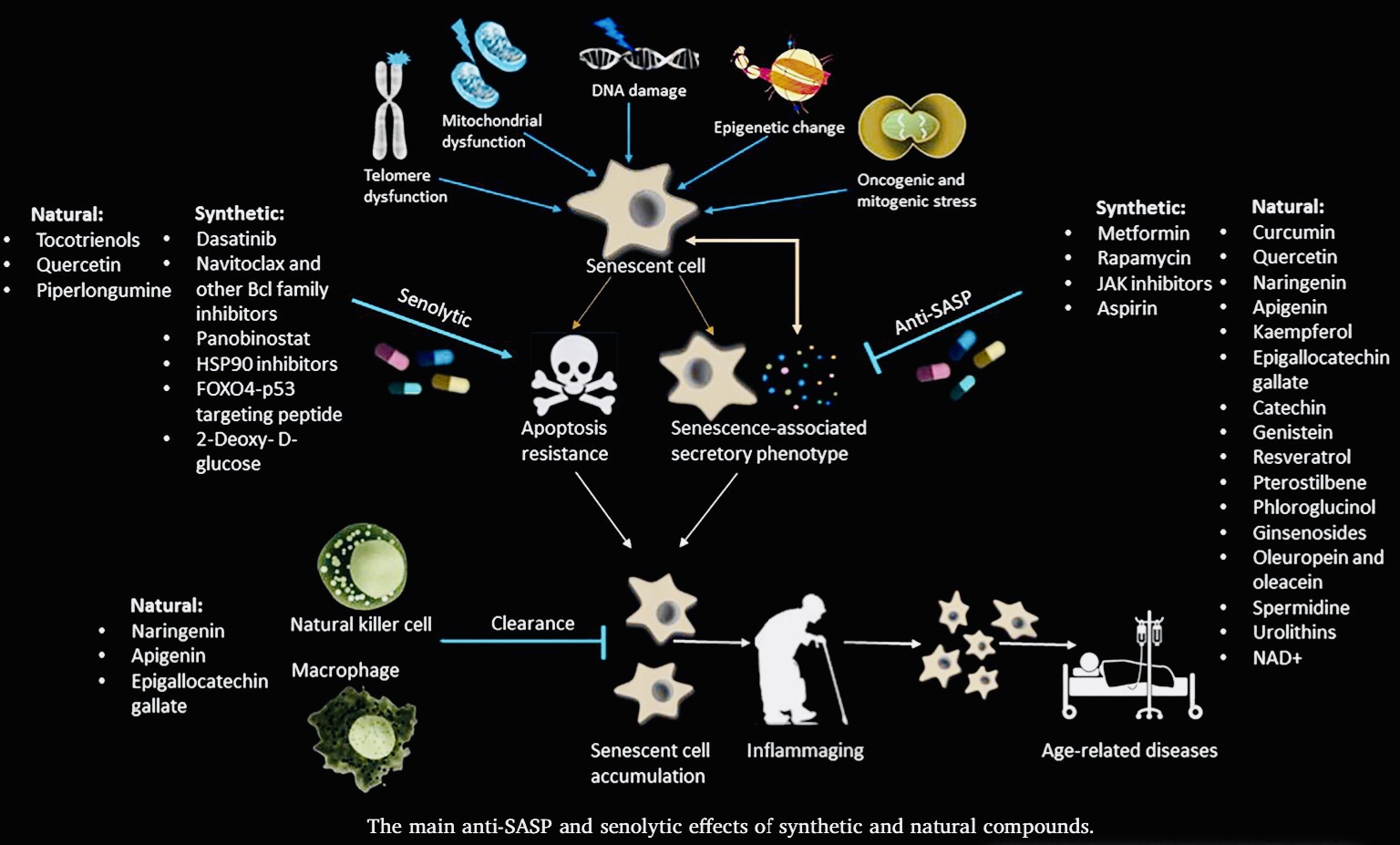




























































don –
Rich Ryan – April 9, 2020
As we age more and more of our cells stop working right, and become Senescent or “Zombie” Cells. They just take up space without doing their jobs, and most of them release toxins into the bloodstream as well. Senolytic blend is meant to enable the body into killing these cells off. Knowing this, I was very excited to try Senolytic blend and boy was I not disappointed!
The first thing I noticed was incredible mental clarity. Then after a week, I had to look twice in the mirror, as I started to look younger. I felt less hungry, and more vibrant and alive. Then, and I kid you not, I started winning money at the casino! I’ve been playing around with my intuition and roulette for years, and when I started to take big doses of Senolytic (1/2 tsp twice a day), my intuition sharpened and I started to hit more numbers! To date, I’ve made triple my money back on Senolytic. Not a bad deal eh? Time to order more!! 🙂 🙂 🙂
In my opinion, this is the best blend Interstellar has come out with yet. Really really impressive!!
don –
Teresa Kuhn – April 11, 2020
WOW! WOW! WOW! These new blends are the bomb! Gavin, I own every blend you sell except the brand new Anti-Adipogenic. In the last few weeks I added Gynostemma, Helicobacter and Senolytic, and it’s made a huge difference. I started taking your blends last summer, developed major FOMO so bought every one you offer, and I have experienced more energy, less appetite, thicker hair, better focus but nothing compares to the energy and focus that I’ve got today. Fasting for 24 hours is a breeze. Thank you Gavin, you are brilliant, and I am so grateful for all you do and your committment to helping others achieve amazing health and well being.
don –
Alex Tice – May 5, 2020
I Am Riding Senolytic Right Now…
⭐️⭐️⭐️⭐️⭐️
As Above, So Below,
The first step to make sure you are preventing a zombie apocalypse on the outside of you is to halt and clear the zombie apocalypse on the inside of you. With this blend, you can kiss the cellular self-sabotage goodbye. In experiencing this Senolytic Blend, I have never felt more like myself ever. We all know this life is a journey where we will continue to travel up our own personally self-constructed ladder to heaven some call Destiny. What they don’t tell us is that the ladder is built inside of us on a cellular, DNA level of spiritually conscious cosmic tapestry that we are. Gavin and the herbs of Nature are helping us go right at the Heart of Healing, tapping into our Innate Unlimited Potential. The Sacred Self-Generating Sanctum is beyond our current imaginative capacity and straight into the Remembrance of what it means to be Human: True Diamond Sun Angelic Unified Broadband Spiritual Stands of Life Eternal.
Symphonic Stardust Expansion.
Gavin is a master of his craft. From purity and quality of selection, to exceptional formulation and design with kyrstalized precision, packaging and professional delivery and service; in every part of the process Gavin and the Interstellar Team leave their mark and open inspiration deep within all participants choosing to go on the inward voyage of a lifetime. As one applies and experiments with the framework that Interstellar has pre-trailblazed for us all, connecting and developing a living relationship with the herbs and the healing forces of Nature comes easier and goes deeper… way deeper !!!
This Senolytic blend will take you through the complete circle of evolution and then up-step you to the next level of your Core Identity and mission purpose of mind, body, and spiritual unfoldment. Whatever you came here to do. Whatever you cant rest from until you become your own living example of Indelible Human Potential Actualized in Full-Empowerment, know that the blends are here as a manifestation of your Eternal Power, and whenever you engage with them, you are in essence taking your power back and returning order, balance, and harmony within the universe. Everything emanates from within, you are your own Source of Power and Truth, you are the Epi-Center and this Senolytic Blend will irrevocably light your fire to StarShine in Authenticity of Your True Eternal Nature.
don –
Stephen Shuman (verified owner) – May 21, 2020
Wow!!! This blend is a game changer. I have been using Trinity for months now, and I was worried that maybe I had developed a tolerance to it. The first month on Trinity was amazing, but than I hit a plateau with the results. I would do a fast, but then as soon as I came back onto eating, even if it was on an intermittent fasting keto diet, it was a real struggle to lose the extra weight, especially my spare tire.
I started taking the Senolytic blend a little over two weeks ago with my morning Trintiy in coffee, and have continued intermittent keto and suddenly not only am I losing weight again, but I have a TON more energy for everything in my life!. Not only that, but most of the weight loss is from my gut and love handles. I am down 14.4 lbs so far. Best of all, I am noticing changes in my my body that I was not expecting. My focus is razor sharp. Sex with my girlfriend has not been this good in months. I have been diagnosed with psoriasis, and my skin has not looked this good in years!!
-Stephen Shuman
don –
Mychael Maldonado (verified owner) – May 22, 2020
I started takin Gavins blends awhile back and was recommended the sample pack
Which had a variety of different blends I could try out. I tried all of them and my favorites were Peel and Spice and Trinity.
Also Matcha and Pine pollen.
Alot of these blends have very good benefits to your body in a kind of miraculous way.
Its like your body saying WHAT THE HECK WAS THAT! !
In a way like Please give me more.
Every herb and every flower and every bark that is in these herbs treat and feed every cell in your body that is damaged and rebuilds them so that you FEEL BETTER, LOOK BETTER, THINK BETTER, LOVE BETTER..
THEY are amazing and worth your attention.
Peel and Spice have me looking
10 years younger….
Trinity has me and my mind at Peace.
This NEW BLEND SENOLYTIC
Is a BOX Office Smash Hit
It clears your FOG and gives you Mental Clarity, Energy, Focus,
It FEEDS your SOUL
IT IS THE REAL DEAL so to say.
And I combine it with the rest.
It even gives me more endurance and strength…
Its the needed Cherry on top of the whole Cake….thank you
Gavin for your knowledge and help that we all need.
But never make a jump towards
Because we are BLINDED in are way of thinking..and limited…
But with these blends there are no Limitations …
There are no I CAN’TS
ONLY I WILL AND I CAN.
Take that Leap Now……
You won’t have any regrets….
don –
Sebastian Gaeta – June 20, 2020
This was the first interstellar blend I have tried. I didn’t really know what to expect with this. Im in my early thirties and generally very healthy and take a lot already, so I didn’t know if I would notice anything. I wasn’t sure how many senolytic cells I had or what the difference would feel like. That said I barely took any of the ingredients listed here before so I was intrigued. I wasn’t fully fasting but trying to be as keto as possible.
I started to really look forward to taking it, I found there was a new level of clarity I was finding myself in most days without realising. Also I found synchronicities increasing drastically and strange situations and items manifesting, sometimes quite literally large items that I had been looking for endlessly with no success turning up on my doorstep. Looking back some very big plans have been set in motion in this period and important projects finished. Things are just coming together quite effortlessly where as some were dragging on for months with no resolution before.
Very interested to see if this continues from where it left off when I try this again or if this effect diminishes now I’ve finished my supply!
Either way definitely a new convert to Interstellar! It looked the part from the ingredients list and certainly is.
don –
Rochelle Vitler (verified owner) – September 6, 2020
I started using Interstellar Blends about nine months ago and since then I can honestly say I have not had a single day where I’ve felt sick or had any sort of sniffle, cough or cold. Every day I am mentally alert, calm and physically healthy.
About three months ago I added Senolytic Zombie Cell Killer to the daily mix of Trinity, Super Hair, Autophagy and either Nebula or Thermo.
Senolytic seems to have cleared up a niggle in my left knee and a dodgy lower back that I’d accepted as part of getting older. Neither gives me any trouble anymore. Fasting is also easier than ever.
The most exciting difference for me with Senolytic though has been that my 50-year-old body, which was flabby from losing 35 kg probably a bit too quickly (even though I exercised the whole time) is starting to tone up. I thought that might not happen and was prepared to accept the weight loss and deal with the soft, squishy body but the changes are exciting and very much welcome. I’ll certainly be keeping Senolytic in the mix.
Thanks Gavin for these awesome blends.
don –
Debra Ferguson (verified owner) – September 7, 2020
I’ve been dealing with health issues for 7-8 years now. When they started, I was around 35 – lifting weights and jogging every day, feeling like I was aging like a fine wine to…. breaking my health somehow. It most likely due stress at the time. I was crazy Type A personality but have worked diligently on my mind and spirit since then so now I’m just as cool as the Fonz 24-7. But I’ve never really gotten back to full health. I’ve seen 17 doctors since then. My symptoms are in line with auto immune issues and NO ONE CAN TELL ME WTF is WRONG.
I have chronic swelling of the hands/fingers, hairloss, joint pain in my hips, mystery skin issue on my torso that isn’t visible but feels like really tiny, rough patches of skin, muscle tightness and pain sometimes so severe even having a 4 hour deep massage doesn’t bring it relief, mystery rash (peri oral dermatitis) that comes and goes, inflammation in my esophagus, sleep issues and even though I exercise daily, my calves have this weird rippling like cellulite but it’s not? I don’t know. It’s weird.
Ultimately, I think the chronic inflammation is at the root cause of all my issues as I know when it’s so rampant in your body, your cells cannot perform. But after 38 different labs in the last 5 or so years, the only thing that shows up is high eosinophils levels which can be linked to allergies or parasites (been tested for both – negative – and even treated for parasites regardless). The levels are not SO high that it sets off the alarm bells though. Everything I mentioned above sometimes leaves me depressed and ready to call it a life. The chronic pain is exhausting. I don’t take meds. Just deal with it.
I’ve been to a gastroenterologist, dermatologist, rheumatologist, naturopath, functional medicine practitioner, DUMB ASS FUCKING REGULAR WESTERN MEDICINE DOCTORS… I’ve done every diet know to man, including a 30 day water fast. Ketosis is definitely something that allows me to feel “better” but still not 100%. I will not give up until I get my health back. If something doesn’t work, I will try something else. Most recently, I did a genetics test and it showed that I’m predisposed to have a hard time creating the elements necessary to detox the body. My body struggles to create glutathione and its precursors as well as with autophagy. I always feel more energetic when I fast and that really explains why.
As you can imagine with all this stuff going on, I’ve had to empower myself with knowledge of all kinds of things because useless doctors can’t help. I used to follow Cole and the Snake Diet but wanted to learn more about dry fasting particularly and that’s when I stumbled upon Gavin’s site. I was, ummm, blown away by the information and after going down the rabbit hole for 2.5 hours, felt like I had leveled up riding a rainbow unicorn towards some new solutions for what’s ailing me. Gavin makes Cole look like romper room. I immediately unfollowed the Snake Diet and signed up for the Interstellar FB group.
Anyhoo – I started with Spice and Peel based on Gavin’s recommendation to treat inflammation, some Hypnotic to address the sleep issues and a sample of the Nebula to see if it would affect my overall mood/mindset. I kind of lack confidence and wanted to give no fucks all the way around. The Hypnotic had me sleeping through the night and feeling rested WITHOUT taking any other non-natural sleep aids. The Nebula certainly had me feeling just relaxed and comfortable in my own skin. Very hard to explain. Like I always have this negative storyline running in the back of my mind and that went away. The Peel and Spice didn’t seem to have as strong as an effect on my inflammation as I had hoped but I also didn’t get the 200:1 concentration which will be my next purchase.
After seeing some measurable results and doing more research on Gavin’s site, I decided to try the Helico. I don’t have stomach issues (very grateful for that) but a few of the studies I read seemed like it couldn’t hurt to try it. I also decided to order the Autophagy Activator and Senolytic based on my genetic report data. Here’s what I experienced:
I usually hop on my treadmill from anywhere from 25-45 minutes. Due to the health stuff, I don’t really have a lot of energy. I work from home and it’s easy to be sedentary. I power through what I can. After taking the combo of Gavin’s blends, my walks went from 25-45 minutes to a minimum 60 minutes to 90. It’s like, even if I want to give up, I’m so focused, I can’t. THIS IS NUTS! I love it. Today, I even increased the difficulty and got my heart rate up about 20 BPM from my normal walks and still did over an hour. Unbelievable. I had less inflammation in my hands when exercising (it usually gets really bad). And I don’t know if it’s related but I had a crazy vision of breathing in golden light and exhaling black air. Almost like whatever negative energy or dysfunction/dis-ease I’m experience was being replaced with white light energy. I can’t really explain it, but it definitely happened. The weird skin issue on my torso is mostly healed. No rash on my face.
I’ve only been taking the Autophagy and Senolytic for 2 days as I’m doing a 7 day fast! Can’t wait to see what the rest of the week looks like! I am telling everyone about the blends. I’m sure I sound like a lunatic but that’s ok. This is the longest testimonial I’ve ever written. If you’ve made it this far, congratulations and buy some blends. You won’t be sorry!